КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
ГЛАВА 5
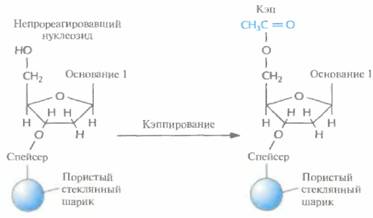
| Рис. 5.6. Кэппирование. Свободные 5'-гидроксильные группы непрореагировавших в первом цикле детритилированных нуклеозидов ацетилируют, чтобы предотвратить их участие в следуюшем цикле. |
лоты или щелочи. Поэтому фосфиттриэфир окисляют с помощью йодной смеси до более стабильного пятивалентного фосфаттриэфира (рис. 5.7). Затем промывают колонку и повторяют весь цикл (детритилирование, активация и присоединение, кэппирование, окисление; рис. 5.1). Все описанные операции проводят до тех пор, пока к растущей цепи в соответствии с программой не присоединится последний нуклеозид. Синтезированные олигонуклеотиды связаны со стеклянными шариками; каждый фосфаттриэфир несет метильную группу; каждый гуанин, цитозин и аденин содержит защищенную аминогруппу, а на 5'-конце последнего нуклеотида находится ДМТ -группа.
Метильные группы удаляют с помощью химической обработки непосредственно в реакционной колонке. Затем отсоединяют олигонуклеотиды от спейсерной молекулы вместе с 3'-гидроксильным концом и элюируют их из колонки; далее последовательно удаляют бензоильные, изобутирильные и ДМТ-группы. 5'-конец цепи фосфорилируют ферментативным (полинуклеотидкиназа Т4+АТР) или химиче-

| Рис, 5.7. Окисление. Фосфиттриэфир окисляется до пятивалентного фосфаттриэфира, что приводит к стабилизации фосфодиэфирной связи и делает ее более устойчивой к действию кислот и шелочей. ДМТ - диметокситритильная группа, Me — метильная группа. |
Химический синтез, определение нуклеотидной последовательности и амплификация ДНК 85
ским методом. Эту реакцию можно проводить и тогда, когда олигонуклеотид еще связан с носителем, но после детритилирования.
Чтобы выход продукта был достаточно высок, эффективность присоединения нуклеотидов на каждом этапе должна быть не ниже 98%, Эффективность контролируют спектрофотометрическими методами, определяя количество удаляемых тритильных групп. Если, например, при синтезе 20-членного олигонуклеотида эффективность каждого цикла равна 99%, то 82% (т, е. 0,9920 · 100) олигонуклеотидов будут иметь именно такую длину. Если же синтезируется 60-членный олигонуклеотид, то при той же эффективности только 55% олигонуклеотидов будут содержать по 60 нуклеотидов. А если средняя эффективность цикла не превышает 98%, то доля олигонуклеотидов заданной длины будет гораздо ниже (табл. 5.1). Фирмы — изготовители коммерческих ДНК-синтезаторов обычно гарантируют среднее значение эффективности присоединения 98%. Но для этого необходимо использовать реагенты и химикаты очень высокой степени чистоты, что не всегда удается выполнить. Как правило, реальная эффективность присоединения составляет 95%, хотя иногда удается достичь и 99%-ной эффективности. Чтобы получить олигонуклеотиды заданной длины, первичные продукты большинства химических синтезов необходимо очистить с помощью либо высокоэффективной жидкостной хроматографии под высоким давлением с обращенной фазой, либо электрофореза в полиакриламидном геле. Поскольку все «неудачные» последовательности короче, чем тот олигонуклеотид, который хотят получить, сделать это нетрудно.
Таблица. 5.1. Средний выход олигонуклеотидов заданной длины (л) при разных значениях средней
эффективности цикла
| Эффективность, % | Средний выход, % | |

| ||
Применение синтезированных олигонуклеотидов
Олигонуклеотиды, синтезированные химическими методами, находят широкое применение в молекулярной биотехнологии. Их используют в качестве зондов при ДНК-гибридизации, линкеров, соединяющих разные молекулы ДНК в экспериментах по клонированию, праймеров при секвенировании ДНК или осуществлении сайт-специфического мутагенеза клонированных генов-мишеней,
1. Нуклеотидную последовательность специфических олигонуклеотидных зондов (длиной 20—40 звеньев) находят из данных об аминокислотной последовательности соответствующих белков.
2. Для получения линкеров синтезируют олигомеры, которые представляют собой палиндромные одноцепочечные нуклеотидные последовательности, спаривающиеся (гибридизующиеся) между собой. Линкеры содержат сайты узнавания для рестрицирующих эндонуклеаз, что позволяет осуществлять с их помощью клонирование фрагментов ДНК (рис. 5.8, А и Б). Короткий дуплекс длиной 6—12 пар нуклеотидов лигируют по тупым концам с ДНК-мишенью (обычно кДНК). Разрезают новую молекулу нужной рестрицирующей эндонуклеазой и получают фрагменты с выступающими одноцепочечными концами (липкими концами), с помощью которых встраивают ДНК-мишень в соответствующий вектор. Прежде чем проводить встраивание, рестрицированную смесь фракционируют для отделения ДНК с липкими концами от лишних линкерных молекул. Вектор тоже обрабатывают рестриктазой, отжигают его с фрагментами ДНК с липкими концами и сшивают с помощью ДНК-лигазы фага Т4. ДНК-мишень не должна содержать сайтов рестрикции, присутствующих в линкерной последовательности, в противном случае она также будет расщепляться ферментом.
3. Один из вариантов линкерных последовательностей, так называемые «адаптеры», час-
86 ГЛАВА 5

|
| Рис. 5.8. Типичные линкеры и адаптер. А, EcoRI-линкер, состоящий из 6 пар нуклеотидов. Б. EcoRI-линкер из 8 пар нуклеотидов. В. BamHI-SmaI-адаптер с BamHI-липкими концами и сайтом узнавания для SmaI. |
то содержат сайты для двух и более рестрицирующих эндонуклеаз (рис. 5,8, В). С их помощью можно встраивать кДНК в вектор лигированием по тупым концам, а затем вырезать, используя другую рестриктазу, Адаптор, изображенный на рис. 5.8, В, встраивают в BamНI-сайт вектора перед включением ДНК-мишени в SmaI-сайт по тупым концам. После клонирования кДНК вырезают из вектора с помощью рсстриктазы BamHI. В этом случае вектор не должен содержать SmаI-сайтов, и ни вектор, ни кДНК не должны нести ВаmHI-сайтов,
4, Одноцепочечные олигонуклеотиды из -17—24 звеньев используют в качестве праймеров при секвенировании ДНК и проведении ПЦР.
5. Одноцепочечные олигонуклеотиды используют в качестве праймеров для сайт-специфического мутагенеза in vitro.
6. Необходимость в химическом синтезе нуклеотидной последовательности, кодирующей какой-то конкретный белок, может возникнуть тогда, когда клонирование соответствующего гена затруднено. При этом нуклеотидную последовательность гена находят из данных об аминокислотной последовательности белка. К химическому синтезу прибегают и тогда, когда кодоны, из которых состоит данный ген, плохо считываются организмом-хозяином, и уровень трансляции оказывается очень низким. В таком случае можно синтезировать ген с таким набором кодонов (оптимизация кодонов), при котором аминокислотная последовательность кодируемого белка остается прежней, а кодоны считываются хозяйским организмом более эффективно.
Синтез генов
Если химически синтезированную двухцепочечную ДНК предполагается использовать в качестве гена или его фрагмента, то каждую из ее цепей синтезируют отдельно. Получить короткие гены (60—80 п. н.) технически несложно: для этого синтезируют комплементарные цепи и затем отжигают их. В случае крупных генов (>300 п. н.) приходится применять специальные стратегии, поскольку эффективность каждого цикла химического синтеза никогда не достигает 100%, Например, если ген состоит из 999 пар нуклеотидов, а эффективность каждого цикла равна 99%, то доля полноразмерных одноцепочечных ДНК по окончании процесса составит не более 0,004%. Чтобы решить эту проблему, синтетические (двухцепочечные) гены собирают из модулей — (одноцепочечных) фрагментов длиной от 20 до 100 нуклеотидов.
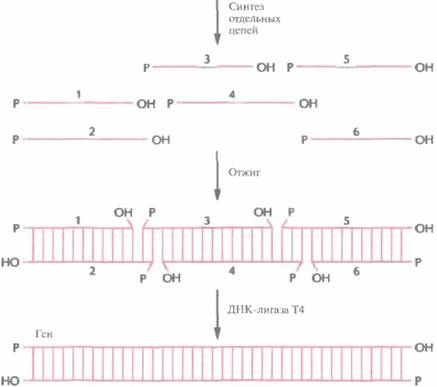
Один из способов конструирования синтетического гена заключается в получении набора олигонуклеотидов длиной 20—60 нуклеотидов каждый с перекрывающимися концами. Нуклеотидные последовательности цепей задают так, чтобы после отжига концевые сегменты гена имели тупые концы. Каждый внутренний сегмент имеет выступающие 3'- и 5'-концы, комплементарные таковым соседнего сегмента (рис. 5.9). После сборки гена остается сшить од-
Химическийсинтез,определение нуклеотидной последовательности и амплификацияДНК 87
|
| Рис. 5.9. Сборка синтетического гена из коротких олигонуклеотидов. Синтезируют отдельные олигонуклеотиды длиной от 20 до 60 звеньев каждый с такими нуклеотидными последовательностями, чтобы при отжиге из них образовалась двуцепочечная молекула. Оставшиеся одноцепочечные разрывы сшивают с помощью ДНК-лигазы Т4. |
ноцепочечные разрывы с помощью ДНК-лигазы Т4. Синтетические гены могут быть сконструированы так, чтобы помимо белок-кодирующих последовательностей они содержали концевые участки, обеспечивающие их встраивание в клонирующий вектор (сайты для рестри-цируюших эндонуклеаз), а также, если это необходимо, сигнальные последовательности для правильной инициации и терминации транскрипции и трансляции.
Для получения полноразмерных генов другим способом тоже вначале синтезируют специфический набор перекрывающихся олигонуклеотидов длиной от 40 до 100 звеньев. При их отжиге происходит спаривание 6—10 3'- и 5'-концевых взаимно комплементарных нуклеотидов, а между ними остаются большие бреши. Протяженность спаренных участков достаточно велика, чтобы стабилизировать всю структуру. Бреши заполняют ферментативным путем с помощью ДНК-полимеразы I Escherichia coli, использующей 3'-гидроксильные группы для инициации репликации и одноцепочечные участки в качестве матрицы. Оставшиеся одноцепочечные разрывы сшивают с помощью ДНК-лигазы Т4 (рис. 5.10).
Более протяженные гены (>1000 п. н.) обычно собирают из двухцепочечных фрагментов, каждый из которых в свою очередь состоит из 4-6 перекрывающихся олигонуклеотидов (от 20 до 60 п. н, каждый). Если после синтеза и отжига образуется достаточное количество фрагментов, то их просто соединяют друг с другом. В противном случае каждый фрагмент клонируют и амплифицируют. Двуцепочечные фрагменты последовательно соединяют друг с другом до образования полноразмерного гена. Чтобы гарантировать
88 ГЛАВА 5

Рис. 5.10.Сборка протяженного гена in vitro с участием ферментов. Вначале химическими методами синтезируют отдельные олигонуклеотиды с такими нуклеотидными последовательностями, чтобы при отжиге между ними образовывались спаренные участки длиной 6—10 пар нуклеотидов. Оставшиеся между ними бреши заполняют с помощью ДНК-полимеразы I Е. coli, а одноцепочечные разрывы сшивают ДНК-лигазой Т4.
правильность нуклеотидной последовательности химически синтезированного гена, секвенируют каждый двухцепочечный фрагмент, а затем и весь ген.
Методы секвенирования ДНК
Исчерпывающую информацию о молекуле ДНК можно получить, только определив ее нуклеотидную последовательность. Так, секвенировав ген, часто удается установить его функцию, сравнив его нуклеотидную последовательность с таковыми для генов, функция которых уже известна. Без данных о нуклеотидной последовательности невозможно проводить исследования по молекулярному клонированию. Секвенирование того или иного фрагмента ДНК можно провести либо химическим методом, разработанным А. Максамом и В. Гилбертом, либо ферментативным, предложенным Ф. Сангером, но в
Химическийсинтез, определение нуклеотидной последовательности и амплификация ДНК 89

|
| Секвенирование ДНК с помощью терминирующих дидезоксинуклеотидов |
| F. Sanger, S. Nicklen, A. R. Coulson Proc. Nail. Acad. Set. USA 74: 5463-5467, 1977 |
| Специфическая ферментативная амплификация ДНК in vitro: полимеразная цепная реакция |
| К. В. Mullis, F. A. Faloona, S, J. Scharf, R. K. Saiki, G. T. Horn, H. A. Erich Cold Spring Harbor Symp, Quant. Biol. 51: 263-273, 1986 |
| Создание новых методов — это необходимая предпосылка развития любой отрасли науки. Они позволяют получать недоступную прежде информацию, что в свою очередь приводит к более глубокому пониманию сути наблюдаемых явлений и стимулирует дальнейшие исследования, порождающие новые открытия. Что касается молекулярной биотехнологии, то ее основой стали такие мощные методы, как секвенирование ДНК и ПЦР. Определение нуклеотидной последовательности ДНК методом ферментативного копирования с остановкой удлинения цепи, осуществляемого ДНК-полимеразой, — быстрый, относительно простой, недорогой и надежный метод. Помимо того что нуклеотидная последовательность фрагмента ДНК является его исчерпывающей характеристикой на молекулярном уровне, она позволяет также идентифицировать его кодирующую область, подобрать потенциальные праймеры для ПЦР, выявить мутационные изменения в гене. До появления в 1977 г. дидезоксиметода Сангера для секвенирования ДНК использовали метод сайт-специфического химического расщепления цепи (А. М, Maxani, W. Gilbert, Proc. Natl. Acad. Sei. USA 74: 560-564, 1977). А еще | раньше секвенирование нуклеиновых кислот сводилось к определению нуклеотидных последовательностей РНК. Для этого нужный фрагмент ДНК сначала транскрибировали в РНК с помощью РНК-пол имеразы, а затем определяли нуклеотидную последовательность последней. Процедура была весьма сложной и длительной и состояла в следующем: радиоактивно меченную РНК обрабатывали различными рибонуклеазами, затем осуществляли хроматографическое разделение образовавшихся продуктов, повторно обрабатывали их ферментами, проводили щелочной гидролиз продуктов второго расщепления, осуществляли хроматографическое разделение продуктов гидролиза, определяли очередность олигонуклеотидов, основываясь на перекрывании их концевых участков, и воссоздавали исходную молекулу. С появлением дидезокси-метода эта процедура практически перестала использоваться. Теперь секвенируют не саму РНК, а ДНК, синтезированную на РНК как на матрице с помощью обратной транскриптазы, и применяют не метод Максама и Гилберта, а метод Сангера, появившийся после того, как была | создана система клонирования на основе фага M13. Возможность прямого секвенирования ДНК произвела настоящую революцию в исследовании молекулярных основ различных болезней человека и в разработке методов их диагностики и лечения. Огромное влияние на многие области исследований, в том числе и на молекулярную биотехнологию, оказала разработка метода ПЦР (Kary Mullis; U.S. patent 4,683,202), С возможностью получения больших количеств ДНК амплификацией сегментов клонированной или геномной ДНК была решена проблема клонирования ДНК-копий редких молекул мРНК, скрининга геномных библиотек, выявления генных мутаций, физического картирования хромосом и т. д. Первым практическим применением ПЦР было создание тест-системы для диагностики серповидноклеточной анемии (Saîki et al., Science 230: 1350-1354, 1985). ПЦР — это уникальная методика, равных которой нет среди других хорошо известных методов. Начиная с 1986 г. с ее помощью выполнено более 10 000 исследований, и несмотря на их огромное разнообразие, перспективы использования ПЦР представляются еще более впечатляющими. |
настоящее время наиболее широко используется так называемый дидезоксинуклеотидный метод.
Дидезоксинуклеотидный метод секвенирования ДНК
Дидезоксинуклеотид — это полученный искусственным путем нуклеотид, лишенный 2'- и 3'-гидроксильных групп при углеродных атомах сахарного кольца (рис. 5.11, А). У дезоксинуклеотида, входящего в норме в состав ДНК, отсутствует только 2'-гидроксильная группа (рис. 5.11,5). Удлинение цепи во время репликации ДНК происходит в результате присоединения очередного нуклеозидтрифосфата к 3'-гидроксильной груп-
Дата добавления: 2015-04-16; просмотров: 74; Мы поможем в написании вашей работы!; Нарушение авторских прав |