КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Приведите схему установки и опишите порядок проведения эксперимента по определению вольтамперной характеристики жидкого стального электрода в оксидном расплаве.
Схема электрических цепей установки и методика проведения экспериментов
Электрохимическая ячейка, применяемая для поляризационных измерений на жидких электродах, представляет собой корундовую воронку(4), запрессованную в графитовую подставку. В нижней части воронки находится столбик (5) исследуемого жидкого металла (карбонильное железо ОСЧ, ТУ 6-09-3000-78, класс 13-2), отделенного от непосредственного контакта с графитом промежуточным слоем диборида циркония (6), который практически не растворяется в железе и обладает электронной проводимостью. Остальная часть воронки заполнена шлаком (3). Электрический контакт источника тока с исследуемым электродом осуществляется через графитовую подставку (7) и вольфрамовый токоподвод (1). Вспомогательный электрод (2), одновременно являющийся и электродом сравнения, изготовлен в виде плоской спирали из вольфрамовой проволоки. Поляризацию его не учитывали в связи с большой величиной поверхности.
Электрическая схема установки для поляризационных измерений (прилагается). Электрохимическая ячейка для поляризационных измерений на жидких электродах (прилагается).
Катодную поляризацию железного электрода исследовали гальваностатическим методом с компенсацией омического падения напряжения в электролите. В качестве источника постоянного тока использовали УИП – 1. Сопротивления R1, R2 в плечах моста выбирали одинаковыми, на два порядка величины превышающими сопротивление электрохимической ячейки. В этом случае поляризующийся ток равен половине общего в цепи и не зависит от потенциала электрода. Балансировку моста осуществляли переменным резистором R3. Постоянный ток, проходящий через электрохимическую ячейку, подключенную к одному из плеч моста, измеряли многопредельным миллиамперметром МII04 класса точности 0,2. Для нахождения величины поляризации измеряли фактическую (i ¹ 0) и равновесную (i = 0) разности потенциалов между электродами с помощью высокоомного потенциометра ППТВ-1. Нуль-прибором служил осциллограф С1-30. С целью повышения точности измерений вместо одиночных выключений тока использовали его импульсное модулирование с помощью реле. Момент компенсации визуально регистрировали на экране осциллографа по исчезновению разрывов на кривой «потенциал φ – время t». Существенной особенностью данной схемы является возможность постоянного наблюдения кривой «φ - t», что позволяет непрерывно проводить компенсацию с точностью до 0,1 Ома. Однако в отличие от коммутаторного метода, стационарность изучаемых процессов здесь почти не нарушается, т.к. уровень пульсаций тока при работе реле не превышает 15%.
Необходимые значения плотности тока (i) рассчитывали по формуле:
i = I / 1.5*π*(d/2)2,
где I – величина поляризующего тока, d – диаметр застывшего столбика металла после опыта (6…9 мм.). Величина числового коэффициента выбрана равной 1,5, т.к. форма мениска металла оказалась средней между полусферической и плоской.
Для приготовления исследуемого электрода порошок карбонильного железа помещали в нижнюю часть корундовой ячейки на подкладку из диборида циркония, расплавляли в атмосфере аргона и дегазировали в вакууме (P~ 10 Па). Затем металл замораживали, помещали в верхнюю часть воронки кусочки шлака, устанавливали вспомогательный электрод, термопару, разогревали систему до температуры опыта и начинали измерения.
Измерение поляризации (h) проводили потенциометром ППТВ – 1 ( Кл. точности – 2), показания (Δφпот) которого отличаются от истинного значения h на величину ΔUосц = IRнескомпл, фиксируемого на экране осциллографа с точностью ± 0,25 мВ:
h = Δφпот - ΔUосц.
Таким образом, используемая в данной работе усовершенствованная схема гальваностатического метода позволяет с высокой точностью измерять как плотность протекающего через ячейку тока, так и поляризацию исследуемого электрода.
27. В чем принцип ориентационно-размерного соответствия Данкова? Связано ли с ним явление зональности при восстановлении железной руды? (см. Климов 98%)
Сначала рассматриваем принцип превращения Байкова.
Восстановление оксидов идет от высших к низшим, через все устойчивые оксиды,
Fe2O3→Fe3O4→FeO→Fe
V2O5→VO2→V2O3→VO→V
Рассмотрим как будет восстанавливаться кусок руды

Если восстановитель твердый углерод, то контакт точечный, который не обеспечивает нормального протекания процесса.
Поэтому процесс восстановления идет через газовую фазу
С+1/2О2→СО
Fe2O3+CO→Fe3O4+CO2 – процесс восстановления
CO2+C=2CO – регенирация газового восстановителя( топлива)
Сначало молекула СО должна адсорбироваться, а потом уже идет взаимодействие. Потом образуется раствор продукты Fe3O4, он растворяется в гематите, потом раствор становится пересыщенным и далее образуются зародыши новой фазы
Fe2O3+COадсор→Fe2+р-р в Fe2O3+CO2
Eэнергия зародыша =1/3 σS, затраты энергии больше на образование зародыша, времени требуется много поэтому зародышей мало.

В инкубационном периоде скорость роста новой фазы маленькая
Граница раздела фаз облегчает процесс образования новых фаз, так как идет достраивание новой решетки, скорость увеличивается.
 Граница раздела фаз начинает сжиматься, такой процесс носит автокатолитический характер, катализатор появляется во время реакции. (Граница фаз является катализатором).
Граница раздела фаз начинает сжиматься, такой процесс носит автокатолитический характер, катализатор появляется во время реакции. (Граница фаз является катализатором).
Процесс зарождения протекает тем легче, чем ближе структура новой фазы по отношению к материнской.

Если кусок руды распилить, то получиться следующая картина

В этом куске руды можно выявить зоны с разными степенями окисления –зональность.
28. В чем сущность адсорбционно-автокаталитической теории восстановления твердых оксидов газообразными восстановителями? Как можно использовать адсорбционный характер процесса восстановления для изменения скорости процесса? (см. лекции Климов 137-140)
29. При каких условиях возникает зональность в куске восстанавливаемой газами железной руды? В каких системах при восстановлении зональность проявляться не будет? (см. Климов 98%)
Если кусок руды распилить, то получиться следующая картина
В этом куске руды можно выявить зоны с разными степенями окисления – зональность.
Зональность проявляется:
А) Наличие устойчивых промежуточных соединений
Б) Последовательность прохождения реакции при восстановлении( поливалентный материал)
Зональность не проявляется:
А) Простой оксид( Ag2O)
Б) если промежуточный оксид улетает( SiO2+ C→ SiO↑ + CO2)
30. Если в системе находятся в равновесии СО, СО2 и Feтв,, то сколько параметров системы можно менять произвольно? Как изменится это число, если в равновесной системе будут СО, СО2,, FeO и Fe? (см. Климов 98%)
Определяем кол-во термодинамических параметров для задания состояния системы.
Fe+CO2=CO+FeO для реакции при температуре выше 845 К.
2 По правилу фаз Гиббса:
С=k+2-ф, где к-число веществ минус число независимых реакций,
2- температура и давление, ф – количество фаз.
Для данной реакции рассмотрим 2 случая

1 случай: Fe может окисляться до FeO( температура больше 845К)
Fe+CO2=CO+FeO
С=4+2-3=3.
С=3- это значит что независимо можно менять 3 параметра( Температура, давление, и состав)
2 случай:Fe не может окисляться до FeO (температура меньше 845К)
С=3+0-2=1
С=1 – это значит что независимо можно менять 1 параметр ( содержание СО)
Осуществите кинетический анализ растворения двухатомных газов в металлах. Рассмотрите стадии растворения газов в металлах. Выделите стадию, определяющую режим процесса и запишите её кинетическое уравнение. Рассмотрите факторы, влияющие на скорость растворения газов в металлах: температура, давление, форма существования того или иного газа в металле, наличие примесей и легирующих элементов. (345-353) Есин, Гельд Ф-Х ч.2
Растворимость газов в металлических расплавах сопровождается диссоциацией молекул на атомы. Поскольку диссоциация протекает в основном в адсорбционном слое на поверхности расплава, то процесс растворения включает доставку молекул из объема газа к границе раздела, адсорбционно-химический акт и отвод образовавшихся продуктов вглубь металлического расплава. Кинетические характеристики сложного процесса, состоящего из ряда последовательных стадий, определяются параметрами той из них, которая наиболее заторможена. Если предельная скорость доставки газовых молекул к границе покрайней мере на порядок меньше скоростей других стадий, то режим процессов считают внешнедиффузионным, а скорость растворения определяется в основном кинетическими параметрами диффузии в газе. Если такой медленной стадией является адсорбционно-химический акт на поверхности расплава, торежим взаимодействия – кинетический. Если скорость всего процесса определяется диффузией в расплаве, торежим, взаимодействия внутрuдuффузuонный. При соизмеримых предельных скоростях двух или всех последовательных стадий режим взаимодействия называют смешанным. В этом случае изменение кинетических параметров каждой из лимитирующих стадий влияет на скорость всего процесса.
В установившемся режиме скорости последовательных стадий равны, хотя каждая из них оказывает различное сопротивление процессу. Подобно последовательным звеньям электрической цепи, за долю сопротивления данной стадии при не очень больших отклонениях от равновесия можно принять отношение перепада химического потенциала взаимодействующих веществ на этой стадии  к суммарному его изменению на всех стадиях, т. e.
к суммарному его изменению на всех стадиях, т. e.  . В общем случае можно характеризовать сопротивление отношением изменения скорости всего процесса
. В общем случае можно характеризовать сопротивление отношением изменения скорости всего процесса  с концентрацией к изменению предельной скорости данной стадии (
с концентрацией к изменению предельной скорости данной стадии ( 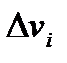 ), т. е. величиной отношения
), т. е. величиной отношения  . Чем выше это отношение, тем больше сопротивление соответствующей стадии i.
. Чем выше это отношение, тем больше сопротивление соответствующей стадии i.
В простейшем случае, когда процесс состоит из двух последовательных стадий: диффузии и химической реакции первого порядка, доля сопротивлений выявить легко.
Выразим скорость диффузии (плотность потока вещества j) равенством, моль/(см2 · с).
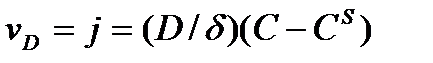 , (1)
, (1)
где D – коэффициент диффузии; δ – толщина диффузионного слоя, в пределах которого принимают, что концентрация линейно уменьшается от значения С в объеме фазы до CS в приповерхностном слое. Скорость химической реакции:
 , (2)
, (2)
где, kx – константа скорости. В установившемся режиме скорости последовательных стадий одинаковы:
 (3)
(3)
Приравнивая правые части соотношений (1) и (2), определяем концентрацию:
 . (4)
. (4)
Подставляя CS в соотношение. (2) и сравнивая полученное выражение с кинетическим уравнением реакции первого порядка
v=kC, (5)
выразим константу скорости процесса k через константы скоростей отдельных стадий:
 . (6)
. (6)
сопротивление сложного процесса
 (7)
(7)
складывается из сопротивлений диффузии RD и химической реакции Rx. Для рассматриваемого случая предельная скорость диффузии (скорость при СS=0)  ,а предельная скорость химической реакции (при СS=С)
,а предельная скорость химической реакции (при СS=С)  . Допустим, что, изменив концентрацию на ΔC, изменили скорость химической стадии на Δvx. Тогда доля сопротивления этой стадии
. Допустим, что, изменив концентрацию на ΔC, изменили скорость химической стадии на Δvx. Тогда доля сопротивления этой стадии
 . (8)
. (8)
сравнивая уравнения (8) и (7), можем заключить, что величина  в данном случае определяет долю химического сопротивления Rx /R. Аналогично можно показать, что отношение
в данном случае определяет долю химического сопротивления Rx /R. Аналогично можно показать, что отношение  равно RD /R. В настоящее время кинетика большинства металлургических реакции изучена недостаточно для количественной оценки сопротивлений в смешанном режиме, что затрудняет оптимизацию процессов. Влияние различных параметров раскрывается четче, когда сопротивление сложному процессу сосредоточено в основном в одной какой-либо стадии, т. е. когда
равно RD /R. В настоящее время кинетика большинства металлургических реакции изучена недостаточно для количественной оценки сопротивлений в смешанном режиме, что затрудняет оптимизацию процессов. Влияние различных параметров раскрывается четче, когда сопротивление сложному процессу сосредоточено в основном в одной какой-либо стадии, т. е. когда  или
или 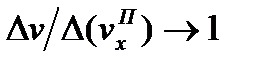 . С учетом этих особенностей рассмотрим кинетические уравнения различных режимов, поскольку в большинстве экспериментальных работ изучали скорости растворения газов, контактирующих с расплавами, по уменьшению их концентраций (парциальных давлений) в закрытых системах; ограничимся обсуждением этого простейшего случая.
. С учетом этих особенностей рассмотрим кинетические уравнения различных режимов, поскольку в большинстве экспериментальных работ изучали скорости растворения газов, контактирующих с расплавами, по уменьшению их концентраций (парциальных давлений) в закрытых системах; ограничимся обсуждением этого простейшего случая.
См. Попель, Сотников, Бороненков. ТМП. Стр.206 -211
Результаты экспериментального исследования скоростей поглощения
Исследованию кинетических характеристик взаимодействия газов с расплавами посвящено сравнительно большое число работ. Однако малая растворимомость газов в жидких, очень малая в твёрдых, сложности анализа и другие экспериментальные трудности снижают точность измерений и не позволяют в настоящее время достаточно точно указать для заданных условий стадию, лимитирующую скорость поглощения того или другого газа и режим дегазации даже вблизи линии ликвидуса.
Интенсивность доставки молекул газа из спокойной среды к поверхности расплава при умеренных давлениях определяется произведением числа молекул в единице объема N насреднюю скорость их движения в данном направлении  см·c-1, где М – молярная масса. Переходя к числу молей в единице объема п=N/NА и считая газ идеальным (п = р/ RT), определим. Предельную скорость доставки его к поверхности, отнесенную к 1 см2, моль/(см2·c):
см·c-1, где М – молярная масса. Переходя к числу молей в единице объема п=N/NА и считая газ идеальным (п = р/ RT), определим. Предельную скорость доставки его к поверхности, отнесенную к 1 см2, моль/(см2·c):
 , ()
, ()
Эта скорость прямо пропорциональна давлению и обратно пропорциональна квадратному корню из молярной массы газа. В частности, при. p=0.1013 МПа и Т=1873 К для водорода и других газов она составляет:
| Газ· · · · · · · · | Н2 | N2 | О2 | СО | СО2 | Н2О |
| и, моль/(см2 ·c) | 0,7 | 0,19 | 0,17 | 0,19 | 0,15 | 0,23 |
Найденные экспериментально скорости поглощения водорода и азота жидким железом в этих условиях гораздо ниже. Они имеют порядок 10-5 - 10-6 моль/(см2·c) и интенсивно возрастают при повышении температуры. Кажущиеся энергии активации процессов абсорбции водорода и азота, по данным различных исследователей, составляют 60 – 80 и 80 – 120кДж/моль соответственно, тогда как диффузия в газе является безактивационным процессом.
Эти и другие особенности процессов позволяют заключить, что при парциальных давлениях > 10 Па диффузия компонента в газе не оказывает заметного влияния на скорости их растворения в железе.
Две другие стадии (адсорбционно-химический акт и массоперенос в расплаве), по-видимому, имеют соизмеримые скорости, и в зависимости от условий опыта каждая из них может оказывать большее или меньшее сопротивление процессу. Так, в ряде работ, проведенных в различных условиях (контакт с газом металла, находящегося в тигле, обдув капли, удерживаемой в газе электромагнитным полем, перемешивание расплава в индукционной печи), установлено замедляющее действие повехностно-активных веществ (ПАВ) – кислорода, серы и др. – на поглощение железом водорода и азота. Например, с увеличением массового содержания кислорода в железе от0,01 до 0,1%, скорость растворения водорода снижается в четыре раза. Занимая часть поверхности, кислород и сера, как капиллярно-активные вещества, затрудняют доступ азоту и водороду для непосредственного взаимодействия с расплавом, вследствие чего замедляется кинетическая стадия исоответственно снижается скорость всего процесса. Вследствие малых концентраций в объёме расплава за пределами адсорбционного слоя (0,01 - 0,04%) эти вещества не оказывают заметного влияния на коэффициенты диффузии и другие характеристики массопереноса в расплаве. Вместе с тем выявленное экспериментально ускорение абсорбции газов с повышением интенсивности перемешивания расплава при неизменной поверхности может быть следствием уменьшения толщины диффузионного слоя. При проведении исследований не всегда удавалось сохранить постоянство поверхности, так как скорость абсорбции возрастала при увеличении интенсивности перемешивания гораздо быстрее, чем для смешанного и даже чисто диффузионного режимов, поэтому роль внутреннего массообмена существенна. Неизбежное присутствие различных, хотя и небольших количеств ПАВ в железе и другие неконтролируемые помехи не позволяют однозначно установить порядок реакции: по одним данным скорость пропорциональна  , по другим –
, по другим –  . Последнее в соответствии с уравнениём (V.53) при
. Последнее в соответствии с уравнениём (V.53) при  подтверждает существенную роль адсорбционно-химического акта в кинетике поглощения. Остановимся на ряде экспериментальных фактов. Выявлено повышение скорости поглощения азота при введении в расплав алюминия и кремния. Причем влияние алюминия оказывается более сильным: на каждую0,1%(моль) А1 в железе скорость при 1873 К возрастает примерно на 3%, а при таких же содержаниях кремния – примерно на 1%. Введение в железо углерода практически не влияет на скорость поглощения водорода и азота. Ускоряющее действие алюминия и кремния связывают частично со снижением концентрации кислорода в расплаве при введении этих элементов, частично с повышением в их присутствии коэффициентов диффузии азота и водорода в железе.
подтверждает существенную роль адсорбционно-химического акта в кинетике поглощения. Остановимся на ряде экспериментальных фактов. Выявлено повышение скорости поглощения азота при введении в расплав алюминия и кремния. Причем влияние алюминия оказывается более сильным: на каждую0,1%(моль) А1 в железе скорость при 1873 К возрастает примерно на 3%, а при таких же содержаниях кремния – примерно на 1%. Введение в железо углерода практически не влияет на скорость поглощения водорода и азота. Ускоряющее действие алюминия и кремния связывают частично со снижением концентрации кислорода в расплаве при введении этих элементов, частично с повышением в их присутствии коэффициентов диффузии азота и водорода в железе.
Выявлено также, что при одинаковых условиях скорость поглощения железом водорода в 8 – 10 раз выше, чем азота. Коэффициенты диффузии этих компонентов в железе при 1873 К различаются почти на два порядка: Dн =(1 – 8) 10-3см2 с, а DN=(4 – 7) 10-5см2 с.
Если допустить, что скорости лимитируются массопереносом веществ в расплаве, то в условиях естественной и регулируемой конвекции они связаны с коэффициентами диффузии приближённым соотношением:
 ()
()
Экспериментально выявленное соотношение скоростей поглощения оказывается близким найденному по формуле (). Константы скоростей адсорбции газов расплавами при этих температурах н6е определены. Также неоднозначны выводы по кинетическим характеристикам поглощения железом кислорода. Высокая энергия активации процесса (130 – 200 кДж/моль), пропорциональность скорости концентрации О2 в газе и сильное замедление при введении небольших количеств серы свидетельствуют о существенной, а может быть и определяющей роли кинетического торможения. Отмечается также, что кислород поглощается гораздо медленнее водорода. Коэффициенты диффузии кислорода в железе определены с большими погрешностями, что затрудняет анализ внутреннего массопереноса.
Таким образом, из рассмотрения опытных данных следует, что абсорбция газов расплавленным железом происходит обычно в смешанном режиме, в котором соизмеримы скорости адсорбционно-химического акта и массопереноса компонента в расплаве. При изменении внешних условий изменяется доля кинетического и диффузионного сопротивлений.
Осуществите кинетический анализ дегазации металлов. Рассмотрите стадии дегазации металлов. Математически опишите стадию, определяющую режим процесса. Рассмотрите факторы, влияющие на скорость дегазации металлов: температура, давление, форма существования того или иного газа в металле, наличие примесей и легирующих элементов. Покажите, что отличает кинетику дегазации металлов от кинетики растворения в них газов. (см. лекции Климов 88-96) (203-215) Попель
При дегазации металлических расплавов повторяются все стадии процесса растворения, только в противоположном направлении и в обратной последовательности, поэтому формально-кинетические уравнения этих процессов аналогичны. Существенно могут различаться только скорости адсорбционно- и десорбционно-кинетических стадий, так как в прямом процессе реализуется адсорбция из газа, а десорбция – в расплав, а при дегазации вещество адсорбируется из расплава и десорбируется в газ. Поскольку в процессах поглощения газов большая, а при сильно развитых гидродинамических потоках определяющая доля сопротивления приходится на кинетические стадии, то вдали от равновесия следует ожидать различия скоростей насыщения и дегазации расплавов. Сказанное подтверждается опытными данными. Многие исследователи утверждают, чтоудаление из расплавленного железа водорода, азота и оксида углерода осуществляется медленнее, чем насыщение его этими газами. Экстракция азота и водорода при перемешивании расплава инертными газами происходит по реакциям второго порядка, т. е.  . При дополнительном перемешивании расплава извлечение водорода ускоряется при увеличении числа оборотов мешалки от 0 до 140 об/мин, тогда как дальнейшее повышение числа оборотовпрактически не влияет на скорость процесса вследствие незначительной доли внутридиффузионного торможения. Замена аргона гелием при продувке расплавов не влияет на скорость экстракции водорода, а замена инертных газов хлором, активно взаимодействующим с водородом, существенно ускоряет процесс. Отмеченные особенности, а также замедляющее действие ПАВ (кислорода и сepы) на экстракцию водорода и азота и сравнительно высокие кажущиеся энергии активации процессов (ЕН ≈100 кДж/моль, EN≈130 – 160 кДж/моль) позволяют заключить, что при продувке расплава инертными газами или при перемешивании другими способами дегазация железа протекает в режиме, близком к кинетическому. Причем скорость всего процесса определяется, по-видимому, соединением адсорбированных атомов в молекулы, поэтому основным уравнением, описывающим скорость дегазации, является, равенство (1).
. При дополнительном перемешивании расплава извлечение водорода ускоряется при увеличении числа оборотов мешалки от 0 до 140 об/мин, тогда как дальнейшее повышение числа оборотовпрактически не влияет на скорость процесса вследствие незначительной доли внутридиффузионного торможения. Замена аргона гелием при продувке расплавов не влияет на скорость экстракции водорода, а замена инертных газов хлором, активно взаимодействующим с водородом, существенно ускоряет процесс. Отмеченные особенности, а также замедляющее действие ПАВ (кислорода и сepы) на экстракцию водорода и азота и сравнительно высокие кажущиеся энергии активации процессов (ЕН ≈100 кДж/моль, EN≈130 – 160 кДж/моль) позволяют заключить, что при продувке расплава инертными газами или при перемешивании другими способами дегазация железа протекает в режиме, близком к кинетическому. Причем скорость всего процесса определяется, по-видимому, соединением адсорбированных атомов в молекулы, поэтому основным уравнением, описывающим скорость дегазации, является, равенство (1).
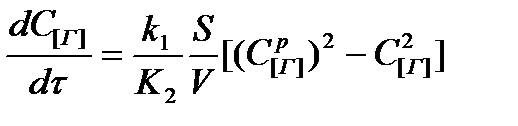 . (1)
. (1)
Сравнить численные значения констант скоростей удаления различных газов не представляется возможным, так как во многих работах они включают наряду с кинетической константой k1 и другие постоянные величины, стоящие в правой части уравнения (1), значения которых обычно не раскрываются. Для азота, например, при 1873 К приводятся значения константы скорости 0,7 – 1,1 см/(% · с), что позволяет оценить прядок скорости дегазации при различных значениях удельных поверхностей.
Опишите кинетику окисления углерода, растворённого в металле, кислородосодержащими газообразными веществами. Рассмотрите стадии процессов и факторы, влияющие на каждую из них. Опишите, как можно выявить режим процесса. Объясните причины появления критических концентраций. (см. лекции Климов 96-103) (215-222 и 301-310) Попель
Взаимодействие кислорода и других окислительных газов (СО2, Н2О) с углеродом, растворённым в железе, имеет место во всех сталеплавильных процессах, где расплавленный металл в большей или меньшей степени вступает в контакт с газовой фазой. Продуктами реакции также являются газы: в основном монооксид углерода, а при малых концентрациях углерода в металле частично и СО2. Обладая ничтожной растворимостью в металле, молекулы монооксида углерода десор6ируются с реакционной поверхности в газовую фазу. При развитии реакции в глубине расплава СО обособляется в газовые пузыри, которые, всплывая в расплаве, перемешивают его. Перемешивание способствует прогреву металла, выравниванию концентраций компонентов. Наряду с этим в пузыри СО поступают из расплава растворенные газы (Н2, N2) и удаляются в атмосферу, поэтому термодинамическому и кинетическому анализу взаимодействия углерода, растворенного в железе, с окислительными газами уделяется очень большое внимание.
Схема процесса окисления углерода.
Трёхфазная система газ/шлак/металл в условиях сталеплавильной печи неравновесна. Химический потенциал кислорода оказывается наибольшим в газовой фазе и наименьшим в металле, поэтому кислород переходит из газовой фазы через шлак в металл.
Молекулы О2 за счёт конвективной диффузии доставляются из газовой фазы к шлаку, где происходит адсорбция молекул и их диссоциация (распадение на атомы)
 , (1)
, (1)
затем происходит ионизация адсорбированного кислорода и переход Fe из двухвалентного в трёхвалентное состояние
 (2)
(2)
Это приводит к обогащению поверхностного слоя шлака ионами  и обеднению катионами Fe2+. Ионы диффундируют от границы газ/шлак к границе шлак/металл. Достигая границы шлак/металл, анионы восстанавливаются при взаимодействии с металлом
и обеднению катионами Fe2+. Ионы диффундируют от границы газ/шлак к границе шлак/металл. Достигая границы шлак/металл, анионы восстанавливаются при взаимодействии с металлом
 . (3)
. (3)
Осуществляется переход кислорода из шлака в металл по реакции
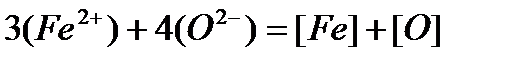 . (4)
. (4)
Затем кислород и углерод диффундируют в металле к месту реакции. Эта реакция гетерогенная.
 . (5)
. (5)
Образование пузырька СО в объёме металла маловероятно. Пузырьки зарождаются на подине. Пузырёк растёт за счёт постоянного подвода [C] и [О] к границе раздела пузырёк/металл. После отрыва пузырька на подине остаётся газовая линзочка. Происходит «донное кипение». Когда подина ошлакована, зарождение пузырьков газа идёт на границе металл/шлак. Пузырьки образуются мелкие и в большом количестве, что приводит к вспениванию шлака, снижению его теплопроводности.
 . (6)
. (6)
Будем считать р{CO}=const
 (7)
(7)
f[C], f[O] – коэффициенты активности,
[C], [O] – концентрации реагентов.
Скорость окисления углерода возрастет, пропорционально его концентрации в расплаве при малых содержаниях, а при больших не зависит от нее. Эта (критическая) концентрация, при которой изменяется вид кинетического уравнения, снижается с повышением интенсивности ввода окислителя, с повышением, температуры и зависит отприсутствия третьих компонентов в расплаве.
Правильный выбор лимитирующего звена процесса обезуглероживания может быть сделан на основе теории критических концентраций С.И.Филиппова. Он принимал за критическую концентрацию углерода при протекании процесса обезуглероживания 0,2 – 0,3 %. Величина критической концентрации определяется интенсивностью подвода окислителя к металлу. При концентрациях углерода выше критической процесс обезуглероживания лимитируется скоростью подвода окислителя, т. е. диффузией кислорода в металле. Скорость обезуглероживания не зависит от концентрации углерода, а зависит от факторов, определяющих подвод кислорода. При концентрациях углерода ниже критических скорость обезуглероживания определяется диффузией углерода.
Величина критической концентрации углерода определяется строением поверхностного слоя на границе раздела шлак – металл или газовый пузырь – металл. Так как углерод и кислород являются поверхностно-активными элементами и их концентрация в поверхностном слое превышает концентрацию в объеме металла, то при снижении концентрации углерода в металле при его окислении концентрация кислорода в поверхностном слое становится больше по сравнению с углеродом и процесс будет определяться уже доставкой углерода к месту реакции.
На скорость обезуглероживания оказывает влияние и стадия образования пузырька СО. Поскольку растворимость СО в металле бесконечно мала, то для осуществления реакции взаимодействия углерода с кислородом необходимо, чтобы образующаяся при этом СО выделилась в новую фазу. Формой существования такой фазы является пузырек СО в объеме металла или газовая атмосфера над металлом.
Опишите кинетику окисления компонентов из расплавленного железа кислородсодержащими газообразными веществами. Рассмотрите стадии процессов и факторы, влияющие на каждую из них. Опишите, как можно выявить режим процесса, объясните причины появления критических концентраций.
1. Подвод молекул газа к поверхности металла V1=(D{r(г)}/δr)(Cv{r(г)}-Cω{r(г)}).
2. Адсорбция молекул газа на поверхности металла V2=kадс(1-θ).
3. Диссоциация газовых молекул на атомы V3=kхCω{r(г)}.
4. Десорбция атомов газа в сторону металла V4=kдесθ.
5. Отвод (диффузия) газа в объем металла V5=(D[r]/δM)(Cv[r]-Cω[г]).
Для определения лимитирующей стадии сравнивают константы скорости
V  д=(D/δ)(Cv-Cω), Vx=kхCω, Vд=Vx, (D/δ)(Cv-Cω)=kхCω, Cω=((D/δ)Cv)/(kх+D/δ)=βCv/(β+kx), β – коэф. массопереноса.
д=(D/δ)(Cv-Cω), Vx=kхCω, Vд=Vx, (D/δ)(Cv-Cω)=kхCω, Cω=((D/δ)Cv)/(kх+D/δ)=βCv/(β+kx), β – коэф. массопереноса.
V=Vд=Vx=kхCω=kхβCv/(kх+β)=Cv/(1/kх+1/β), V=Cv/Rр-ии,
Rр-ии=1/kх+1/β=Rд+Rx, Rр-ии=∑Ri – соответствующие стадии.
kэф=kxβ/(kx+β), 1/kэф=1/kx+1/β, kx>>β, kэф=β.
Режимы процесса: внешний диффузионный, внутренний диффузионный, кинетический. См 7, 8, 9
Дата добавления: 2015-04-18; просмотров: 69; Мы поможем в написании вашей работы!; Нарушение авторских прав |