КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Моделирование сахарного диабета
Для углубленного изучения этиологии и патогенеза СД используются различные экспериментальные модели. Первая модель СД была получена в 1889 г. О. Минковским и Дж. Мерингом, которые вызвали диабет у собак путем удаления поджелудочной железы и установили, что необходимым фактором для развития СД является недостаточность секреции инсулина.
Парентеральное введение аллоксанакрысам вызывает некроз β-клеток островков и через 24-48 ч приводит к развитию гипергликемии. При аллоксановом диабете снижено содержание инсулина в крови и поджелудочной железе, повышена чувствительность тканей к экзогенному инсулину, снижено содержание гликогена в печени, отмечается жировое истощение и снижается синтез белка, т.е. экспериментальная картина соответствует развитию СД 1 типа.
Экспериментальный диабет у кроликов получают путем внутривенного введения дитизона.При остром течении такого диабета развиваются высокая гликемия (>50 ммоль/л), глюкозурия, гиперкетонемия, полиурия, полидипсия, уменьшение массы тела. Животные погибают в течение 5-10 дней при состоянии, напоминающем диабетическую гипергликемическую кому.
Введение собакам СТГв больших дозах вызывает соматотропный (гипофизарный) диабет с классическими симптомами - ги-
пергликемией, глюкозурией, полиурией и повышенным содержанием свободных жирных кислот (СЖК) в крови. В начальный период при использовании такой модели концентрация инсулина в 15-20 раз превышает нормальный уровень, затем нормализуется и в дальнейшем падает в результате истощения функциональных резервов инсулярного аппарата.
Введение глюкокортикоидовв больших дозах приводит к развитию стероидного диабета, для которого характерна начальная гиперинсулинемия, снижение чувствительности к инсулину (инсулинорезистентность) и последующее поражение инсулярного аппарата с возникновением классического синдрома инсулинодефицитного диабета.
В современной классификации сахарного диабета (ВОЗ, 1999) выделяют 4 группы: СД 1 типа (аутоиммунный и идиопатический), СД 2 типа, гестационный и другие специфические формы сахарного диабета.
Сахарный диабет I типа (деструкцияβ-клеток, обычно ведущая к абсолютному дефициту инсулина)
Иммуноопосредованный диабет.Основным звеном патогенеза СД 1 типа является воспалениев островках Лангерганса, приводящее к деструкции β-клеток и дисфункции остальных клеточных типов островка (это эндокриноциты энтодермального происхождения - α-, β-, δ- и PP-клетки; кровеносные капилляры, симпатические терминали, эфференты и афференты блуждающего нерва, нейроны, глиальные клетки и резидентные тканевые макрофаги). Это воспаление имеет аутоиммунный характер (аутоиммунный инсулит) и вызывает гибель β-клеток преимущественно путем апоптоза
(рис. 12-23).
СД 1 типа может быть результатом дефекта системы иммунологического надзора, связанного с наследованием определенной комбинации генов главного комплекса гистосовместимости. Показана четкая ассоциация заболевания с HLA-генами - В8 и В15 (гены I класса комплекса гистосовместимости); генами DR3 и DR4; DQ B1 и DQ B2 (гены II класса комплекса гистосовместимости). В связи с этим определение данных генов у больных СД 1 типа и у членов их семей может служить генетическими маркерамизаболевания. Так, самый высокий титр антител к инсулину отмечен у лиц с антигеном В15, а у 95% больных потенциальный риск развития
 Рис. 12-23. Этиологические и патогенетические факторы развития сахарного диабета (СД) 1 типа.HLA (human leukocytes antigen) - человеческий лейкоцитарный антиген
Рис. 12-23. Этиологические и патогенетические факторы развития сахарного диабета (СД) 1 типа.HLA (human leukocytes antigen) - человеческий лейкоцитарный антиген
заболевания обусловлен сочетанием антигенов DR3+DR4. В пользу генетической предрасположенности этой формы диабета свидетельствует сочетание ее с другими аутоиммунными заболеваниями (болезнь Грейвса, тиреоидит Хасимото, целиакия, витилиго, болезнь Аддисона, ревматоидный артрит). Таким образом, в основе СД 1 типа лежит генетически обусловленный дисбаланс отдельных популяций иммунокомпетентных клеток.
Маркерами иммунной деструкцииβ-клетокявляются аутоантитела к антигену β-клетки (ICA), аутоантитела к инсулину (IAA), аутоантитела к глутаматдекарбоксилазе (GADA) и аутоантитела к тирозин-фосфатазам IA-2 и IA2b. Один вид, а обычно и более, этих аутоантител присутствует у 85-90% индивидуумов при первоначальном обнаружении гипергликемии натощак.
Т-лимфоциты играют важную роль в инициации и развитии аутоиммунного процесса, приводящего к уничтожению инсулинсинтезирующих клеток. Основная роль принадлежит цитотоксическим Т-лимфоцитам. Роль Т-звена в формировании диабета показана в опытах на мышах линии NOD (Non-Obese Diabetic mice - диабетические мыши без ожирения со спонтанно развивающимся аутоиммунным диабетом), которых с помощью тимэктомии и введения антител к молекулам CD4+ защищали от развития спонтанного аутоиммунного диабета.
Метаболическими маркерамиСД 1 типа является уровень инсулина и С-пептида в крови и моче (в норме соотношение концентраций инсулина и С-пептида равно 1/3); гликозилированного гемоглобина - HbA1c (более 5% от всего содержания гемоглобина); нарушение толерантности глюкозы в ГТТ.
Патогенез деструкцииβ-клетокподжелудочной железы достаточно сложен. В последние годы установлено, что существенную роль в этих механизмах играет свободный радикал - оксид азота (NO), который образуется из L-аргинина под действием нейрональной или эндотелиальной NO-синтазы и участвует в передаче межклеточных сигналов (снижает тонус периферических сосудов с помощью инсулинопосредованного эндотелийзависимого механизма вазодилатации; регулирует процессы в нервной ткани). Существует третья изоформа NO-синтазы - индуцибельная (iNOS),которая появляется в клетках при повышении содержания провоспалительных цитокинов и может выделяться непосредственно в β-клетку, вызывая цитотоксические и цитостатические эффекты.
Каскад иммунньгх реакций, приводящих к деструкции β-клеток, инициируется также резидентными макрофагами островков Лангерганса. Активированный диабетогенными факторами макрофаг продуцирует провоспалительные цитокины - интерлейкин (IL) 1, фактор некроза опухолей (TNF) α, интерферон γ, способные разрушать β-клетки. Примером одного из механизмов деструкции служит экспрессия гена iNOSв β-клетке с помощью IL-1, что увеличивает образование оксида азота. Важно отметить, что ген iNOSлокализуется на 11-й хромосоме рядом с геном, кодирующим синтез инсулина, что позволяет предполагать вовлеченность этих двух областей в патогенез СД 1 типа.
Значимость NO как одного из ведущих патогенетических факторов деструкции β-клеток подтверждена применением никотинамида и аминогуанидина. Эти препараты ингибируют экспрессию iNOS,уменьшают образование NO и предотвращают деструкцию β-клеток и манифестацию СД 1 типа.
Начальное повреждение β-клеток уменьшает количество ГЛЮТ 2 на их цитоплазматической мембране, снижая чувствительность к глюкозе и тем самым уменьшает продукцию инсулина еще до их выраженной деструкции. Клинически СД 1 типа манифестирует (у детей чаще кетоацидозом), когда повреждены 80-90% островковых клеток. При раннем обнаружении деструкции β-клеток с помощью перечисленных выше маркеров и при адекватном лечении повреждение клеток можно предупредить и остановить. Лечение иммунодепрессантами (циклоспорин А) до развития гипергликемии может существенно снизить вероятность заболевания СД 1 типа.
Многие больные с СД 1 типа становятся зависимыми от инсулина и находятся в состоянии риска по кетоацидозу. На последней стадии заболевания секреция инсулина минимальна или отсутствует, что проявляется низким или неопределяемым уровнем С-пептида плазмы. Иммуноопосредованный СД обычно начинается в детском и подростковом возрасте, но может развиться в любой период жизни, даже у пожилых людей 80-90 лет.
Идиопатический диабет.Некоторые формы СД 1 типа не имеют известной этиологии. Больные с идиопатическим диабетом имеют постоянный дефицит инсулина и наклонность к кетоацидозу, но у них отсутствуют маркеры иммунной деструкции и нет связи с HLA- генами.Эта форма имеет четкое наследование, механизм которого еще неясен. Идиопатический диабет чаще всего развивается у лиц
африканского или азиатского происхождения. У пациентов с этой формой диабета эпизодически выявляются кетоацидоз и различные степени инсулинодефицита. Причиной инсулиновой недостаточности в этом случае может быть и частое употребление в пищу продуктов, содержащих цианиды (корни маниока, сорго, ямс). Цианиды в нормальных условиях обезвреживаются при участии серосодержащих аминокислот, но при белковой недостаточности, повсеместно распространенной у лиц, проживающих в странах Азии и Африки, создаются условия для накопления цианидов и повреждения β-клеток поджелудочной железы.
Актуальной задачей является диагностика предклинического периода развития деструкции инсулярного аппарата. Это достигается с помощью определения аутоантител (ICA, IAA, GADA) или по оценке инсулинового ответа на внутривенную нагрузку глюкозой (ГТТ). Уже на стадии доклинических проявлений деструкции исчезает ранний пик секреции инсулина на глюкозную нагрузку. Если эти метаболические и иммунные признаки сочетаются с наследственной предрасположенностью (HLA-типирование), то можно с большой вероятностью поставить диагноз деструкции инсулярного аппарата на доклинических этапах СД 1 типа.
Сахарный диабет 2 типа (от преобладающей инсулинорезистентности с относительным инсулинодефицитом до преобладающего дефекта секреции инсулина с инсулинорезистентностью)
СД 2 типа - общее собирательное название гетерогенных нарушений углеводного обмена, имеющих, в отличие от СД 1 типа, другую генетическую основу и разнообразные пусковые механизмы развития. Содержание инсулина в поджелудочной железе и крови может быть нормальным или даже повышенным, поэтому говорят об относительной инсулиновой недостаточности, подразумевая недостаточность метаболических эффектов инсулина в тканях. Это явление обозначают специальным термином - инсулинорезистентность. Инсулинорезистентность (ИР) - это снижение реакции инсулинчувствительных тканей на инсулин при его достаточной концентрации, в результате чего глюкоза не усваивается инсулинзависимыми тканями и развивается гипергликемия.
В основе патогенеза СД 2 типа лежат два ведущих фактора: ИРи дисфункция β-клеток.ИР бывает трех типов: пререцепторная, рецепторная и пострецепторная.
Пререцепторная ИРобусловлена следующими механизмами:
1) мутацией гена инсулина;
2) мутацией генов, контролирующих энергетический обмен в β-клетках и секрецию инсулина.
Рецепторная ИРсвязана с дефектом синтеза, ресинтеза или субстратной аффинности инсулиновых рецепторов на β-клетках и других клетках-мишенях, в основе которых лежат:
1) мутации гена инсулинового рецептора (в настоящее время выявлено более 30 точечных мутаций этого гена);
2) повышенное использование рецепторов инсулина;
3) уменьшение количества рецепторов инсулина на поверхности гипертрофированных адипоцитов;
4) увеличение количества висцеральной жировой ткани, имеющей низкий уровень экспрессии рецепторов к инсулину, что обусловливает ее изначальную ИР;
5) блокирование рецепторов к инсулину антителами. Пострецепторная ИРсвязана с патологией ассоциированных с
рецепторами инсулина тирозинкиназы и глюкозных транспортеров. К этому могут привести:
1) повреждение внутриклеточных посредников передачи инсулинового сигнала (например, мутации генов субстрата инсулинового рецептора - IRS-1);
2) снижение чувствительности ГЛЮТ 2 β-клеток поджелудочной железы к глюкозе;
3) снижение мембранной концентрации и активности ГЛЮТ 4 в мышечной и жировой ткани;
4) снижение чувствительности ГЛЮТ 2 к глюкозе в гепатоцитах;
5) нарушение обмена глюкозы в клетках-мишенях инсулина (например, при мутации гена глюкокиназы, гликогенсинтазы, митохондриального гена при MELAS-синдроме).
Наибольшее значение, вероятно, имеют рецепторная и пострецепторная ИР, которая по механизмам развития может быть первичной и вторичной. Первичная ИРопределяется вышеперечисленными генетическими механизмами. Из-за этих нарушений возникает гиперинсулинемия, которая сначала имеет компенсаторный характер, так как необходима для преодоления ИР тканей и поддержания нормального транспорта глюкозы в клетки. Показано, что самым ранним признаком СД 2 типа является нарушение способности мышечных и жировых клеток реагировать на инсулин. До тех пор пока поджелудочная железа способна увеличивать
секрецию инсулина с тем, чтобы восполнить ИР этих тканей, толерантность к глюкозе остается в норме.
Однако с течением времени β-клеткам не удается поддерживать высокий уровень секреции инсулина, что снижает толерантность к глюкозе и постепенно приводит к развитию СД, т.е. возникает дисфункцияβ-клеток.Несмотря на то что у больных этой формой диабета нормальный или повышенный уровень инсулина, его все же недостаточно для компенсации высокой гликемии, характерной для СД 2 типа. Таким образом, секреция инсулина у этих больных неполноценна и недостаточна для того, чтобы компенсировать ИР, поэтому на определенных этапах лечения СД 2 типа пациентам назначают инсулинотерапию, несмотря на достаточно высокий уровень инсулина в крови.
Вторичная ИРсвязана: 1) с хронической гипергликемией,которая способствует десенситизации β-клеток, проявляющейся снижением их секреторной активности; с ожирением,в результате чего повышается продукция адипоцитами лептина, TNF-α, IL-1, IL-6; 2) с дислипопротеинемией,характеризующейся повышением содержания липопротеинов очень низкой плотности (ЛПОНП), липопротеинов низкой плотности (ЛПНП) и снижением уровня холестерина липопротеинов высокой плотности (ЛПВП), что способствует активации окислительного стресса в эндотелиоцитах и эндотелиальной дисфункции (см. раздел 12.5.2). При развитии вторичной ИР дисфункция β-клеток с преобладанием дефекта секреции инсулина выходит на первый план.
Несмотря на гетерогенность патогенетических факторов, обусловливающих ИР при развитии СД 2 типа, можно выделить 3 уровня нарушений гомеостаза глюкозы:
1. Нарушение механизма узнавания глюкозы β-клетками поджелудочной железыи вследствие этого потеря первой фазы секреции инсулина в ГТТ.
2. ИР инсулинозависимых периферических тканей,что приводит к недостаточному транспорту и метаболизму глюкозы в клетках и гипергликемии, результатом чего является нарушение толерантности к глюкозе в ГТТ.
3. Увеличение уровня глюкозы натощак свидетельствует о повышении продукции ее печенью(вследствие недостатка инсулина и/или избытка глюкагона и катехоламинов).
Таким образом, патогенез СД 2 типа может быть представлен следующей последовательностью событий: первичная ИР и дис-
функцияβ-клеток-> действие диабетогенных факторов-> хроническая гипергликемия-> гиперинсулинемия-> вторичная ИР-> нарастающий относительный дефицит инсулина-> атрофия поджелудочной железы-> абсолютный дефицит инсулина(рис. 12-24).
Из внешних факторов, неблагоприятно влияющих на чувствительность тканей к инсулину, наибольшее значение имеют гиподинамияи избыточное потребление жира(см. раздел 12.5.4). Гиподинамия сопровождается снижением транслокации ГЛЮТ 4 в мышечных клетках и усиливает ИР мышечной ткани (см. рис. 12- 15). Избыточное потребление животных жиров, содержащих насыщенные жирные кислоты, приводит к структурным изменениям мембранных фосфолипидов и нарушению экспрессии генов, контролирующих проведение инсулинового сигнала внутрь клетки (пострецепторные механизмы), что также усиливает ИР на уровне
 Рис. 12-24.Этиологические и патогенетические факторы развития сахарного диабета (СД) 2 типа. ГЛЮТ - глюкозный транспортер, ГТТ - глюкозотолерантный тест, СЖК - свободные жирные кислоты, HLA (human leukocytes antigen) - человеческий лейкоцитарный антиген
Рис. 12-24.Этиологические и патогенетические факторы развития сахарного диабета (СД) 2 типа. ГЛЮТ - глюкозный транспортер, ГТТ - глюкозотолерантный тест, СЖК - свободные жирные кислоты, HLA (human leukocytes antigen) - человеческий лейкоцитарный антиген
всего организма. Гипертриацилглицеролемия, в особенности постпрандиальная, часто наблюдаемая у пациентов с висцеральным типом ожирения, сопровождается избыточным отложением липидов в мышцах, которое нарушает активность ферментов, участвующих в метаболизме глюкозы.
Несмотря на то что транспорт глюкозы в клетки в условиях ИР нарушен, липогенетическое действие инсулинана клетки инсулинозависимых тканей может сохраняться и заключается в следующем:
• инсулин активирует ЛП-липазу адипоцитов,способствуя ее переносу на мембрану эндотелиоцитов кровеносных сосудов, где начинается распад хиломикронов и ЛПОНП на СЖК (они депонируются в жировой ткани в виде триацилглицеролов - ТАГ) и глицерол, поступающий в печень для синтеза новых
ТАГ и ЛПОНП;
• инсулин опосредованно тормозит липолиз в жировой ткани, угнетая гормончувствительную ТАГ-липазу, и ТАГ депонируются в адипоцитах. Именно поэтому СД 2 типа сопровождается ожирением.
Такое действие инсулина на ферменты жирового обмена объясняет развитие при СД 2 типа ретенционной гиперлипемиии дислипопротеинемии IV и V типов,когда в крови увеличивается содержание ЛПОНП и ТАГ (рис. 12-25).
При СД 1 типа отмечается транспортная гиперлипемия(см. раздел 12.5.3), а больные худеют. Кроме того, повышенный липолиз при СД 1 типа усугубляется усилением липолитического эффекта соматотропина, АКТГ, глюкагона, адреналина и тироксина в условиях абсолютной недостаточности инсулина.
Отмечая роль ожиренияв патогенезе СД 2 типа, необходимо подчеркнуть наличие взаимосвязи между СД 2 типа и степенью ожирения. Риск развития данного вида диабета увеличивается в 2 раза при наличии ожирения I степени, в 5 раз - при средней степени ожирения и более чем в 10 раз при наличии III степени ожирения. При этом абдоминальное распределение жира тесно связано с развитием метаболических нарушений (см. раздел 12.5.4).
Представленные сложные взаимосвязи патогенетических факторов, участвующих в механизме нарушений регуляции углеводного и жирового обмена при СД 2 типа, позволили G. Reaven в 1988 г. сформулировать концепцию метаболического синдрома,основными компонентами которого является ИР тканей и гиперинсулинемия, нарушение толерантности к глюкозе и развитие СД 2 типа, абдо-
 Рис. 12-25.Механизм увеличения депонирования жиров в адипоцитах в условиях гиперинсулинемии при СД 2 типа (липогенез > липолиз - увеличение депонирования жира): 1 - активация инсулином ЛП-липазы адипоцитов; 2 - угнетение гч-ТАГ-липазы адипоцитов; 3 - перенос ферментативных комплексов ЛП-липазы на мембрану эндотелиоцитов и связывание их с ХМ и ЛПОНП; 4 - катаболизм ХМ и ЛПОНП; 5 - синтез в печени ЛПОНП из избытка глицерола и СЖК; 6 - избыточное поступление ЛПОНП в кровь; 7 - избыточное поступление СЖК в адипоциты; ТАГ - триацилглицеролы; СЖК - свободные жирные кислоты; ХМ - хиломикроны; ЛПОНП - липопротеины очень низкой плотности; ЛПлипаза - липопротеиновая липаза адипоцитов; гч-ТАГ-липаза - гормончувствительная ТАГ-липаза, - - усиление эффекта; ■ - блокирование эффекта; ® - активация фермента; θ - ингибирование фермента
Рис. 12-25.Механизм увеличения депонирования жиров в адипоцитах в условиях гиперинсулинемии при СД 2 типа (липогенез > липолиз - увеличение депонирования жира): 1 - активация инсулином ЛП-липазы адипоцитов; 2 - угнетение гч-ТАГ-липазы адипоцитов; 3 - перенос ферментативных комплексов ЛП-липазы на мембрану эндотелиоцитов и связывание их с ХМ и ЛПОНП; 4 - катаболизм ХМ и ЛПОНП; 5 - синтез в печени ЛПОНП из избытка глицерола и СЖК; 6 - избыточное поступление ЛПОНП в кровь; 7 - избыточное поступление СЖК в адипоциты; ТАГ - триацилглицеролы; СЖК - свободные жирные кислоты; ХМ - хиломикроны; ЛПОНП - липопротеины очень низкой плотности; ЛПлипаза - липопротеиновая липаза адипоцитов; гч-ТАГ-липаза - гормончувствительная ТАГ-липаза, - - усиление эффекта; ■ - блокирование эффекта; ® - активация фермента; θ - ингибирование фермента
минальное (висцеральное) ожирение, дислипидемия, артериальная гипертензия и атеросклероз (рис. 12-26). Исследования последних лет расширили компоненты метаболического синдрома, добавив к ним гиперурикемию, гомоцистеинемию, гиперандрогению у женщин, нарушения гемостаза (увеличение содержания в крови инги-
 Рис. 12-26.Этиологические, патогенетические факторы и последствия метаболического синдрома
Рис. 12-26.Этиологические, патогенетические факторы и последствия метаболического синдрома
битора активатора плазминогена, гиперфибриногенемия). Наиболее часто в литературе используется термин «метаболический синдром» (метаболический синдром Х), хотя прилагательное «дисметаболический» более точно отражает сущность патогенеза этого синдрома.
В индустриалышх странах распространенность метаболического синдрома среди населения старше 30 лет составляет 10-20%. Метаболический синдром может быть генетически детерминирован, что подтверждается результатами близнецового метода и преобладанием заболеваемости в определенных этнических группах, например, у жителей Индии.
Необходимо особо отметить значение абдоминального (висцерального) ожиренияв развитии ИР тканей, СД 2 типа и метаболического синдрома. Висцеральная жировая ткань, в отличие от жировой ткани другой локализации, непосредственно сообщается с портальной системой печени.При данном типе ожирения активация липолиза в жировой ткани сальника и брыжейки сопровождается избыточным поступлением СЖК в портальную циркуляцию и в печень (выше в 20-30 раз по сравнению с нормой). Висцеральные адипоциты характеризуются высокой плотностью β,,-адренорецепторов, кортикостероидных и андрогенных рецепторов и низким содержанием α2-адренорецепторов и рецепторов к инсулину. Эти особенности определяют высокую чувствительность висцеральной жировой ткани к липолитическому действию катехоламинов и других перечисленных выше факторов и низкую - к липогенетическому действию инсулина (см. раздел 12.5.4).
Таким образом, любые гормональные нарушения, сопровождающие стресс, возрастные гормональные перестройки, вызывают интенсивный липолиз в висцеральных адипоцитах и приводят к выделению большого количества СЖК в портальную циркуляцию и печень. В печени СЖК экранируют инсулиновые рецепторы и препятствуют связыванию инсулина гепатоцитами, обусловливая развитие ИР на уровне печени (снижается транспорт глюкозы в гепатоциты из-за нарушения глюкокиназного механизма, что приводит к гипергликемии), снижается экстракция инсулина гепатоцитами (инсулин становится недоступным для действия инсулиназы гепатоцитов), и развивается системная гиперинсулинемия. СЖК подавляют тормозящее действие инсулина на глюконеогенез, увеличивая продукцию глюкозы печенью. С другой стороны, СЖК в мышечной ткани конкурируют с глюкозой и обусловливают ИР на уровне мышечной ткани, усугубляя гипергликемию (рис. 12-27).
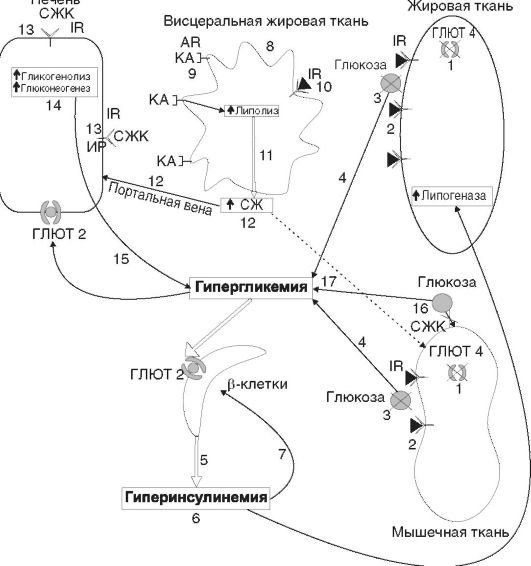 Рис. 12-27.Роль нарушений ГЛЮТ 4 и висцерального ожирения в развитии инсулинорезистентности: 1 - наследственный дефект ГЛЮТ 4; 2 - инсулиновый сигнал не работает (ГЛЮТ 4 остаются в клетках инсулинзависимых тканей); 3 - транспорт глюкозы в клетки инсулинзависимых тканей прекращается; 4 - гипергликемия; 5 - увеличение продукции инсулина β-клетками; 6 - гиперинсулинемия и усиление липогенеза; 7 - истощение β-клеток; 8 - висцеральное ожирение; 9 - увеличение количества β3-адренорецепторов; 10 - снижение количества рецепторов к инсулину; 1l - усиление липолиза в результате действия КА (высокие концентрации КА при действии психогенных факторов); 12 - повышение СЖК в портальной вене печени; 13 - экранирование рецепторов к инсулину СЖК и ИР на уровне печени; 14 - отмена угнетающего действия инсулина на гликогенолиз и глюконеогенез в печени; 15 - гипергликемия натощак; 16 - конкуренция СЖК за поступление глюкозы в мышечные клетки; 17 - гипергликемия
Рис. 12-27.Роль нарушений ГЛЮТ 4 и висцерального ожирения в развитии инсулинорезистентности: 1 - наследственный дефект ГЛЮТ 4; 2 - инсулиновый сигнал не работает (ГЛЮТ 4 остаются в клетках инсулинзависимых тканей); 3 - транспорт глюкозы в клетки инсулинзависимых тканей прекращается; 4 - гипергликемия; 5 - увеличение продукции инсулина β-клетками; 6 - гиперинсулинемия и усиление липогенеза; 7 - истощение β-клеток; 8 - висцеральное ожирение; 9 - увеличение количества β3-адренорецепторов; 10 - снижение количества рецепторов к инсулину; 1l - усиление липолиза в результате действия КА (высокие концентрации КА при действии психогенных факторов); 12 - повышение СЖК в портальной вене печени; 13 - экранирование рецепторов к инсулину СЖК и ИР на уровне печени; 14 - отмена угнетающего действия инсулина на гликогенолиз и глюконеогенез в печени; 15 - гипергликемия натощак; 16 - конкуренция СЖК за поступление глюкозы в мышечные клетки; 17 - гипергликемия
Примечание:IR - инсулиновый рецептор; ГЛЮТ 2 и ГЛЮТ 4 - глюкозные транспортеры; ИР - инсулинорезистентность; СЖК - свободные жирные кислоты; AR - адренорецепторы; КА - катехоламины
При рассмотрении взаимосвязи гиперинсулинемии и артериальной гипертензии выстраивается картина многофакторных взаимоотношений.
У здоровых людей инсулин в физиологической концентрации стимулирует эндотелиальную NO-синтазу через фосфатидилино- зитол-3-киназный путь. В результате увеличивается продукция NO, которая обеспечивает эндотелийзависимую вазодилатацию. В условиях ИР изменяются внутриклеточные сигнальные системы, и трансдукция сигналов через фосфатидилинозитол-3-киназный путь нарушается, с инсулиновых рецепторов сигнал передается через митогенактивируемую протеинкиназу. Активация этого сигнального пути приводит к увеличению продукции эндотелинаи повышению уровня маркеров воспаления и тромбоза.
Кроме увеличения продукции эндотелина, гиперинсулинемия активирует целый ряд механизмов, повышающих тоническое напряжение сосудистой стенки:
• повышение активности симпатических центров головного мозгавследствие усиления метаболизма глюкозы в инсулиночувствительных клетках вентромедиального гипоталамуса;
• увеличение фильтрации глюкозы в почечных клубочках в условиях хронической гипергликемиисопровождается усилением обратной ее реабсорбции вместе с натрием в проксимальных канальцах, приводит к гиперволемии, повышению общего периферического сосудистого сопротивления и увеличению артериального давления;
• повышение уровня ангиотензиногена II,так как в условиях ИР отменяется инсулинопосредованное ингибирование экспрессии гена ангиотензиногена;
• увеличение чувствительности кардиомиоцитов и гладкомышечных клеток сосудов к прессорным воздействиям,так как при ИР и гиперинсулинемии подавляется активность Na+/K+- и Са2+-зависимой АТФаз, что сопровождается увеличением внутриклеточного содержания Na+ и Са2+ и уменьшением концентрации K+ и Mg2+ внутри клеток;
• развитие дислипидемии,которая, способствуя дисфункции эндотелия,приводит к нарушению физиологического механизма NO-опосредованной вазодилатации (см. рис. 12-26).
Рассмотренные выше нарушения углеводного и липидного обмена при метаболическом синдроме и СД 2 типа характеризуются изменениями, приводящими к нарушению микроциркуля-
ции, повышенному тромбообразованию, что является основой для развития осложнений этих заболеваний: раннего атеросклероза и сердечно-сосудистой патологии (ИБС, инфаркт миокарда, инсульт); нефро-, ретино- и нейропатиям; неалкогольному стеатогепатиту (гепатоспленомегалия, фиброз, цирроз); синдрому хиломикронемии (панкреатит, ксантома).
Таким образом, гиперинсулинемия, являясь компенсаторной ответной реакцией организма, направленной на обеспечение адекватного транспорта глюкозы в клетку, одновременно является и патологическим фактором, поскольку вызывает развитие «порочных кругов» в патогенезе метаболического синдрома и СД 2 типа.
Проявления СД 2 типа имеют свои особенности по сравнению с таковыми при СД 1 типа (табл. 12-4).
Таблица 12-4.Основные признаки сахарного диабета 1 и 2 типов
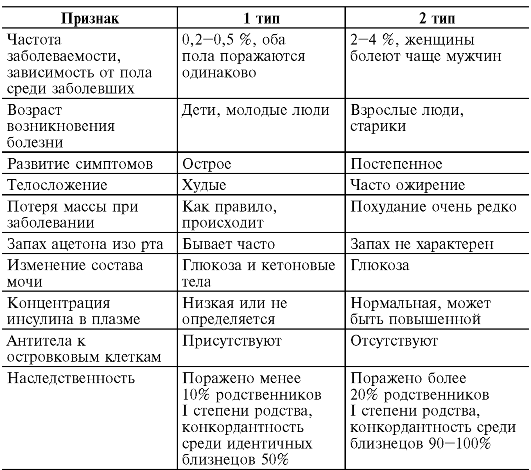
| Ассоциация с HLA | В8, В15, DR3, DR4, DQ B1, DQ B2, Dw3, Dw4 | Нет ассоциации |
| Лечение (основное) | Инсулин | Диета, сульфонилмочевинные препараты |
Гестационный сахарный диабетсвязан с ИР, развивающейся у женщины во время беременности (обычно во II триместре). Под этим названием объединяют любые нарушения толерантности к глюкозе, возникшие впервые во время беременности. Во второй половине беременности значительно возрастает уровень плацентарных гормонов, которые подавляют утилизацию глюкозы тканями матери, чтобы обеспечить поступление достаточного количества глюкозы в фетоплацентарную систему, поэтому у беременных уровень глюкозы в крови после приема пищи выше, чем у небеременных.
Постоянная легкая гипергликемия приводит к физиологической гиперинсулинемии. Во второй половине беременности возникает физиологическая инсулинорезистентность,обусловленная плацентарными гормонами - прогестероном, эстрогенами, пролактином и плацентарным лактогеном. ИР также способствует развитию гиперинсулинемии, поэтому, как правило, диабет беременных по патогенезу сходен с СД 2 типа и поддается диетотерапии. В большинстве случаев диабет беременных проходит после родов, но он существенно может повышать риск развития СД 2 типа у матери в будущем.
Другие специфические формы сахарного диабетавключают в себя очень большую группу патологических процессов, имеющих как первичный, так и вторичный характер нарушений углеводного обмена. Основными причинами генетических дефектов функции β-клеток могут быть мутации гена глюкокиназы (все варианты MODY - от англ. Maturity onset type diabetes - «диабет взрослого типа у молодых лиц»), митохондриальных генов - MELAS- синдром; выработка антител к инсулину и к рецептору инсулина может привести к необычным формам иммуноопосредованного диабета.
Вторичный инсулинодефицитвозникает в результате следующих заболеваний:
• экзокринного отдела поджелудочной железы (хронический панкреатит - алкогольный и тропический, травма, гемохроматоз, неоплазии и др.);
• эндокринопатиях (акромегалия, синдром Иценко-Кушинга, тиреотоксикоз, глюкагонома, феохромоцитома, синдром Конна, гиперандрогенемия и др.); болезнях печени (цирроз, хронический активный гепатит);
• генетических синдромах (синдром Дауна, Клайнфельтера, Шерешевского-Тернера, Прадера-Вилли, хорея Гентингтона и др.);
• вирусных инфекциях (цитомегаловирусная, краснуха и др.);
• при использовании лекарственных препаратов и химических веществ (кортикостероиды, оральные контрацептивы, тиазидовые диуретики, диазоксид, вакор, циклоспорин А, пентамидин и др.).
Алкогольный панкреатитявляется наиболее частой причиной хронического панкреатита. Заболеванию подвергаются люди в основном среднего возраста. Уровень глюкозы в крови весьма неустойчив в связи с нарушениями в диете, ухудшением всасывания и переваривания, снижением эндокринной функции поджелудочной железы; периодически могут возникать гипер- и гипогликемии; кетоацидоз - явление редкое. Лечение ферментами поджелудочной железы улучшает гликемический контроль.
Гемохроматозсопровождается отложением железа в печени, поджелудочной железе, коже, половых органах. Подозрение на наличие гемохроматоза у больного сахарным диабетом должно возникнуть при сочетании бронзового оттенка кожи, гепатомегалии с аномальными функциональными печеночными тестами и импотенцией. Эффективное лечение гемохроматоза флеботомией и железосвязывающими препаратами улучшает толерантность к глюкозе.
Обширные поражения печени,такие как цирроз, снижают экстракцию инсулина печенью из портальной циркуляции, что приводит к периферической гиперинсулинемии и ИР. У предрасположенных пациентов на этом фоне возникает диабет. Нарушение толерантности к глюкозе и диабет средней тяжести отмечаются у 50-80% пациентов с установленным циррозом.
Повышенный уровень контринсулярных гормонов,особенно у предрасположенных лиц, снижает резервную функцию β-клеток и приводит к гипергликемии. Диагностика этих состояний обычно
не вызывает затруднений в связи с классической симптоматикой и клинической картиной, обусловленной избыточной продукцией гормонов.
Некоторые лекарственные препараты(см. выше) могут нарушать толерантность к глюкозе, вызывая либо ИР, либо дисфункцию β-клеток.
Часть редких генетических аномалий(см. выше) может также сопровождаться СД. Несмотря на разнообразие причин, к категории «вторичного» СД клинически может быть отнесена относительно небольшая его часть (<1%). Устранение или лечение основного заболевания у этих пациентов может привести к «излечению» СД.
Дата добавления: 2015-04-15; просмотров: 318; Мы поможем в написании вашей работы!; Нарушение авторских прав |