КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Анемии: этиология, патогенез, классификация, клинические проявления, диагностика, принципы лечения.
Железодефицитная анемия (ЖДА) – гипохромная микроцитарная анемия, развивающаяся вследствие абсолютного уменьшения запасов железа в организме.
Эпидемиология: ЖДА страдают около 200 млн. людей во всем мире; самая частая форма анемий (80%).
Этиология ЖДА:
1. Хронические периодические кровопотери (желудочно-кишечные кровотечения, маточные кровотечения, гематурия, кровоточащие опухоли, донорство и др.)
2. Повышенный расход железа (беременность, лактация, половое созревание, хронические инфекции)
3. Недостаточное поступление железа с пищей
4. Нарушение всасывания железа (синдром мальабсорбции, резекция желудка, высокая энтеропатия и др.)
5. Нарушение транспорта железа (наследственный недостаток трансферина, АТ к трансферрину)
6. Врожденный дефицит железа (нарушение реутилизации железа и т.д.)
Патогенез ЖДА:
В основе заболевания – дефицит железа, в котором выделяют 2 стадии:
а) латентный дефицит – уменьшаются запасы железа в печени, селезенке, костном мозге, при этом снижается уровень ферритина в крови, происходит компенсаторное усиление всасывания железа в кишечника и повышение уровня мукозного и плазменного трансферина; содержание сывороточного железа еще не снижено, анемии нет
б) собственно ЖДА – истощенные депо железа не способны обеспечить эритропоэтическую функцию костного мозга и, несмотря на сохраняющийся высокий уровень трансферина в крови, значительное содержается содержание сывороточного железа, синтез гемоглобина, развивается анемия и последующие тканевые нарушения.
При дефиците железа, помимо анемии, снижается активность железосодержащих и железозависимых ферментов в различных органах и тканях, уменьшается образование миоглобина ® дистрофические поражения эпителиальных тканей (кожи, слизистых ЖКТ, мочевыводящих путей и др.) и мышц (скелетный, миокарда).
Распределение железа в организме: 57-65% – гемоглобин; 27-30% – железо негемовых белков (ферритин, гемосидерин); 8-9% – миоглобин; до 0,5% – железо ферментов (цитохромов и пероксидаз); 0,1% – транспортное железо (с трансферрином).
Классификация ЖДА:
1) по этиологии:
а) хроническая постгеморрагическая ЖДА
б) ЖДА вследствие повышенного расхода железа
в) ЖДА вследстие недостаточного исходного уровня железа (у новорожденных и детей младшего возраста)
г) алиментарная ЖДА
д) ЖДА вследствие недостаточного всасывания в кишечнике
е) ЖДА вследствие нарушения транспорта железа
2) по стадии развития: латентная анемия и ЖДА с развернутой клинико-лабораторной картиной заболевания
3) по степени тяжести: легкая (Hb 90-120 г/л), средняя (Hb 70-90 г/л), тяжелая (Hb ниже 70 г/л).
Клиника ЖДА:
а) общеанемический синдром:
- общая слабость, повышенная утомляемость, снижение работоспособности, памяти, сонливость, головокружение, шум в ушах, мелькание мушек перед глазами, сердцебиение, одышка при физических нагрузках, склонность к ортостатическим обморокам
- бледность кожи и видимых слизистых оболочек (иногда с зеленоватым оттенком – «хлороз»)
- небольшая пастозность в области голеней, стоп, лица, утренние отеки – «мешки» над и под глазами
- синдром миокардиодистрофии (одышка, тахикардия, часто аритмия, умеренное расширение границ сердца влево, глухость тонов, систолический шум на верхушке)
б) сидеропеничекий синдром (синдром гипосидероза):
- извращение вкуса - непреодолимое желание употреблять в пищу что-либо необычное и малосъедобное (мел, зубной порошок, уголь, глину, сырое тесто, фарш, крупу); чаще встречается у детей и подростков
- пристрастие к острой, соленой, кислой, пряной пище
- извращение обоняния - пристрастие к запахам, которые большинством окружающих воспринимаются как неприятные (бензин, керосин, ацетон, запах лаков, красок, гуталина, нафталина и др.)
- выраженная мышечная слабость и утомляемость, атрофия мышц и снижение мышечной силы; императивные позывы на мочеиспускание, невозможность удержать мочу при смехе, кашле, чихании, возможно даже ночное недержание мочи (слабость сфиктера мочевого пузыря)
- дистрофические изменения кожи и ее придатков (сухость, шелушение, склонность к быстрому образованию на коже трещин; тусклость, ломкость, выпадение, раннее поседение волос; истончение, ломкость, поперечная исчерченность, тусклость ногтей; симптом койлонихии - ложкообразная вогнутость ногтей)
- дистрофические изменения слизистых: ангулярный стоматит - трещины, «заеды» в углах рта; глоссит - ощущение боли и распирания в области языка, покраснение его кончика, в дальнейшем атрофия сосочков («лакированный» язык); склонность к пародонтозу и кариесу; атрофические изменения ЖКТ (сухость слизистой пищевода и затруднения, а иногда боли при глотании пищи, особенно сухой - сидеропеническая дисфагия или симптом Пламмера-Винсона; атрофический гастрит и энтерит)
- снижение репаративных процессов в коже и слизистых
- симптом «синих склер» Ослера - синеватая окраска или выраженная голубизна склер (из-за нарушения гидроксилирования пролина и лизина при дефиците железа склера истончается и через нее просвечивается сосудистая оболочка глаза)
- «сидеропенический субфебрилитет» - длительное повышение температуры до субфебрильных величин
- выраженная предрасположенность к ОРВИ, хронизации инфекций (из-за нарушения фагоцитоза)
Диагностика ЖДА:
а) ОАК: снижение содержания гемоглобина и (в меньшей степени) эритроцитов; микроцитоз; ЦП < 0,8 (= 3*Hb / 3 первые цифры числа эритроцитов); нормальные лейкоциты и тромбоциты
б) БАК: тесты феррокинетики:
1) сывороточное железо (СЖ): женщины - норма 11,5-30,4 мкмоль/л, при дефиците железа < 11,5 мкмоль/л; мужчины - норма 13,0-31,4 мкмоль/л, при дефиците железа < 11,5 мкмоль/л
2) общая железосвязывающая способность сыворотки (ОЖСС): женщины – норма 44,8-70,0 мкмоль/л, при дефиците железа > 70,0 мкмоль/л; мужчины – норма 44,8-70,0 мкмоль/л, при дефиците железа > 70,0 мкмоль/л
3) процент насыщения трансферрина железом (%НТЖ): женщины – норма 25-40%, при дефиците железа < 25%, мужчины – норма 25-50%, при дефиците железа < 25%
4) сывороточный ферритин: женщины – норма 10-100 нг/мл, при дефиците железа < 10 нг/л; мужчины – норма 30-200 нг/мл, при дефиците железа < 30 нг/мл
Принципы рациональной терапии ЖДА:
1. Основа лечения – препараты солевого железа per os; ЖДА невозможно купировать только диетой, богатой железом, т.к. из пищи в тонкой кишке всасывается 2-2,5 мг железа в сутки, а из препаратов – в 10-15 раз больше.
NB! Перед применением препаратов железа следует исключить сидероахрестическую (железонасыщенную) анемию, при которой дефицита железа нет, но оно не используется; образуются гипохромные эритроциты, а железо захватывается клетками макрофагальной системы и откладывается в органах и тканях, вызывая их гемосидероз. Назначение препаратов железа при данной анемии только ухудшит состояние больного!
2. Этапность лечения – 2 этапа (3-5 мес.):
1) купирование анемии (от начала терапии до нормального уровня Hb – обычно 4-6 недель)
2) «терапия насыщения» - восполнение депо железа в организме (8-12 недель - по 30-60 мг ЭЖ/сут).
3. Правильный расчет лечебной и профилактической дозы по элементарному железу (ЭЖ): лечебная доза рассчитывается по содержанию элементарного железа в препарате и для взрослого весом 70-80 кг составляет 100-200 мг ЭЖ
Основные препараты железа для перорального приема: «Ферроплекс»: 1 таблетка = 10 мг ЭЖ, «Ферроцерон»: 1 таблетка = 40 мг ЭЖ, «Феррокальм»: 1 таблетка = 44 мг ЭЖ; пролонгированные формы (1-2 раза/сут): Ферро-градумет»: 1 таблетка = 105 мг ЭЖ; «Мультирет»: 1 таблетка = 105 мг ЭЖ; «Тардиферон»: 1 таблетка = 80 мг ЭЖ; «Сорбифер»: 1 таблетка = 100 мг ЭЖ.
4. Лечение сочетают с одновременным применением аскорбиновой кислоты (0,3-0,5 г на прием), которая в 2-3 раза повышает всасывание железа в кишечнике; целесообразны также антиоксиданты и витамин В6.
5. Оптимально принимать препарат железа за 30 мин до еды, при плохой переносимости – через 1 час после еды, не разжевывая, запивая водой, можно фруктовыми соками без мякоти, но не молоком (кальций молока тормозит всасывание железа); для детей можно использовать сиропы - «Ферринсол», «Гемофер», «Интрофер».
6. Парентерально препараты железа используются по показаниям: синдром мальабсорбции; резекция желудка; резекция верхнего отдела тонкой кишки («Феррум-лек», «Эктофер», «Фербитол»).
7. При уровне гемоглобина < 70 г/л показано переливание эритроцитарной массы.
8. Критерии излеченности:
а) повышение уровня ретикулоцитов на 5-7 день от начала ферротерапии
б) повышение уровня гемоглобина с 3 (и раньше) недели лечения и восстановление его к 6-ой
в) нормализация показателей СФ, СЖ, ОЖСС, %НТЖ по окончании курса лечения
9. Профилактический курс ферротерапии – приём препарата по 30-40 мг ЭЖ/сутки в течение 4-6 недель (тардиферон по 1 таблетке в 2 дня и др.)
Мегалобластные анемии (МА) – группа анемий, основной причиной которых является дефицит фолиевой кислоты и витамина В12 и связанное с этим нарушение синтеза ДНК (при этом страдают прежде всего быстро обновляющиеся ткани – кроветворная и эпителий ЖКТ).
Морфологически при МА деление клеток замедляется, цитоплазма созревает нормально, в результате клетки становятся крупными, содержание РНК превышает ДНК; эритропоэз неэффективный, т.к. на уровне костного мозга образуются мегалобласты – предшественники эритроцитов, разрушающиеся в самом костном мозге.
Классификация мегалобластных анемий:
а) В12-дефицитная анемия
б) Фолиеводефитиная анемия
в) МА, обусловленная наследственным дефицитом ферментов, участвующих в синтезе пуриновых и пиримидиновых оснований (дигидрофолиевой редуктазы, формиминотрансферазы и др.)
г) МА, поддающаяся лечению витамином В1 (врожденное аутосомно-рециссивное заболевание)
д) МА, обусловленная дефицитов витамина С (он участвует в метаболизме фолиевой кислоты)
е) острая мегалобластная болезнь (быстрое развитие МА вследствие действия различных причин – закиси азота, гемодиализа, длительного парентерального питания и др.)
Этиология мегалобластных анемий:
1. Причины дефицита витамина В12:
а) недостаточное поступление его с пищей
б) нарушение его всасывания при недостатке внутреннего фактора Касла (на уровне желудка), поражении дистальных отделов ileum, конкурентном поглощении витамина В12 при дифиллоботриозе, применении некоторых лекарственных средств (аминосалициловая кислота, неомицин)
2. Причины дефицита фолиевой кислоты:
а) недостаточное поступление с пищей (особенно у алкоголиков, подростков, грудных детей)
б) период повышенной потребности в фолиевой кислоте (беременность, грудной возраст, злокачественные опухоли, гемолитическая анемия, гемодиализ)
в) нарушение всасывания (болезни тонкого кишечника: целиакия, спру; применение некоторых ЛС: барбитураты, фенитоин)
г) нарушения метаболизма фолиевой кислоты (применение ингибиторов дигидрофолатредуктазы - метотрексата, триметоприма; алкоголь; недостаточность дигидрофолатредуктазы) и др.
Патогенез мегалобластных анемий:
Фолиевая (птероилглутаминовая) кислота – источник для человека – зеленые овощи и фрукты, минимальная потребность – 50 мкг/сут (при беременности увеличивается в несколько раз), резерв в организме 5-20 мг (половина резерва содержится в печени); в пище находится в конъюгированной форме в виде полиглютамата; основная функция - перенос метильной или формильной группы от одного вещества к другому.
Метаболизм фолиевой кислоты: полиглютамат + γ-глютамилгидролаза в просвете кишечника ® моноглютамат ® всасывание в проксимальном отделе тонкой кишки ® попадание с помощью переносчика в клетку ® потеря метильной группы (при участии витамина В12) ® повторное образование полиглютамата ® использование на нужды клетки.
При дефиците фолиевой кислоты нарушается синтез ДНК в кроветворных клетках из-за нарушения синтеза пуриновых и пиримидиновых оснований.
Витамин В12 (кобаламин) – источник для человека – продукты животного происхождения (мясо, молоко, яичные желтки); минимальная потребность – 2,5 мкг/сут, резерв в организме: 2 мг в печени + 2 мг в других тканях (поэтому дефицит развивается лишь через 3-6 лет после прекращения поступления в организм);
Метаболизм кобаламина: высвобождение кобаламина из пищи в желудке + желудочный R-белок ® комплекс В12+R-белок поступает в ДПК ® соединение с внутренним фактором Касла (вырабатывается париетальными клетками желудка) ® расщепление образовавшегося комплекса в энтероцитах концевого отдела подвздошной кишки ® связь кобаламина с транскобаламином II (переносчиком) ® перенос кровью к костному мозгу и печени.
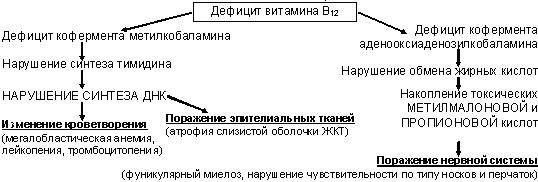 Атрофия желудка с нарушением образования внутреннего фактора Касла и развитием МА – болезнь Адиссона-Бирмера (пернициозная анемия, в 60% при ней обнаруживают АТ к париетальным клеткам желудка или к внутреннему фактору Касла)
Атрофия желудка с нарушением образования внутреннего фактора Касла и развитием МА – болезнь Адиссона-Бирмера (пернициозная анемия, в 60% при ней обнаруживают АТ к париетальным клеткам желудка или к внутреннему фактору Касла)
Клиническая картина В12-дефицитной анемии:
а) поражение пищеварительной системы – чаще всего самые ранние признаки заболевания:
- снижение или отсутствие аппетита, ощущение тяжести и полноты в подложечной области после еды, отрыжка съеденной пищей и воздухом, боль и жжение в языке, в области десен, губ, иногда в области прямой кишки (из-за глоссита, атрофического гастрита и атрофии слизистой кишечника)
- воспалительно-атрофические изменения слизистой полости рта и языка (слизистая рта бледная, с признаками афтозного стоматита; гладкий «лакированный» язык с атрофированными сосочками, потрескавшийся, с участками воспаления ярко-красного цвета, иногда с изъязвлениями - глоссит Hunter)
- пальпаторно - неинтенсивная боль в эпигастральной области, иногда – гепатоспленомегалия
б) поражение кроветворной системы – чаще всего ведущие признаки заболевания:
- общеанемический синдром (как при ЖДА)
- кожа бледная, часто с лимонно-желтым оттенком (в связи с гипербилирубинемией из-за гемолиза), легкая иктеричность склер
- несколько одутловатое лицо, часто – пастозность в области голеней и стоп
- синдром миокардиодистрофии (тахикардия, экстрасистолия, небольшое расширение границ сердца влево, приглушенность тонов сердца, негромкий систолический шум в области верхушки)
в) поражение нервной системы – фуникулярный миелоз – возникает при тяжелом и длительном течении болезни, характеризуется поражением задних и боковых столбов спинного мозга с демиелинизацией и дегенерацией нервных волокон в спинном мозге и спинномозговых нервах:
1) при преимущественном поражении задних столбов:
- жалобы на слабость в ногах, особенно при подъеме по лестнице, при быстрой ходьбе, ощущение ползания мурашек по ногам, онемение ног; больным кажется, что они не чувствуют при ходьбе опоры под ногами, что нога наступает не на твердую землю, а на что-то рыхлое, мягкое, как вата (больные неоднократно как бы «пробуют землю ногой»)
- снижение сухожильных рефлексов, атрофия мышц нижних конечностей
- нарушение функции тазовых органов (недержание мочи, недержание кала)
2) при преимущественном поражении боковых столбов:
- нижний спастический парапарез с резким повышением сухожильных рефлексов и тонуса мышц нижних конечностей
- нарушение функции тазовых органов (задержка мочеиспускания и дефекации)
Клиническая картина фолиеводефицитной анемии отличается от В12-дефицитной анемии отсутствием поражения пищеварительной и нервной системы.
Диагностика В12-дефицитной анемии:
1. Лабораторные исследования:
а) ОАК: гиперхромная (ЦП > 1,1) макроцитарная анемия, анизоцитоз (разная величина эритроцитов, наряду с макроцитами имеются эритроциты нормальных размеров), пойкилоцитоз (изменение формы эритроцитов);
тельца Жолли, кольца Кебота, базофильная пунктация (остатки ядра в мегалоцитах); лейкопения, нейтропения, эозинопения, относительный лимфоцитоз, гиперсегментированные нейтрофилы (большие сегментоядерные нейтрофилы с полисегментированным ядром); умеренная тромбоцитопения
б) миелограмма (пункцию необходимо делать до начала лечения!): раздражение красного кроветворного ростка, его гиперплазия; клетки красного ряда преобладают над клетками белого ряда, отношение лейкоциты/эритроциты 1:2 - 1:3 (при норме 3:1-4:1); мегалобластный тип кроветворения (гиганская клетка с эксцентрично расположенным ядром без ядрышек, нежной хроматиновой сетью) с преобладанием в разгар болезни базофильных мегалобластов («синий костный мозг»); изменение клеток миелоидного ряда; нарушение созревания мегакариоцитов
в) БАК: признаки гемолиза (неконьюгированная гипербилирубинемия, умеренное повышение СЖ)
г) тест Шеллинга - позволяет оценить всасывание витамина В12 в кишечнике в присутствии гастромукопротеина или без него и сделать заключение о патогенетическом варианте В12-дефицитной анемии:
1) Шеллинг – I: больному дают принять внутрь витамин В12, меченный 60Со и через 1-6 часов в/м вводят «ударную дозу» немеченого витамина В12 для насыщения печеночного депо, затем измеряют содержание радиоактивного витамина В12 в суточной моче; снижение его экскреции указывает на нарушение всасывания витамина В12 в кишечнике
2) Шеллинг – II: повторяется тест Шеллинг-I с использованием гастромукопротеина, меченого радиоактивным кобальтом; повышение экскреции радиоактивного витамина В12 указывает на дефицит гастромукопротеина в качестве основного механизма развития В12-дефицитной анемии; если экскреция радиоактивного витамина В12 не увеличилась, причина развития МА - нарушение всасывания витамина
В12 в кишечнике.
2. Инструментальные исследования: ФГДС с биопсией (атрофические изменения слизистой ЖКТ); исследование желудочной секреции (резкое уменьшение количества желудочного сока, ахилия) и др.
Диагностика фолиеводефицитной МА:
а) ОАК – те же признаки, что и при В12-дефицитной анемии
б) миелограмма – те же признаки, что и при В12-дефицитной анемии; при окраске пунктата по Кассу (ализариновым красным) мегалобласты окрашиваются только при В12-дефицитной анемии и не окрашиваются при фолиеводефитиной анемии
в) БАК: признаки гемолиза (неконьюгированная гипербилирубинемия, умеренное повышение СЖ)
г) проба с гистидином - больной принимает 15 г гистидина, после чего определяется экскреция с мочой формиминглутаминовой кислоты за 8 ч после приема гистидина; в норме основная часть гистидина превращается при участии фолиевой кислоты в глутаминовую кислоту, с мочой выводится от 1 до 18 мг формиминглутаминовой кислоты; при фолиеводефицитной анемии выделение формиминглутаминовой кислоты значительно увеличивается ( до 1500 мг)
Лечение МА:
При В12-дефицитной анемии - препараты витамина В12 (цианокобаламин, оксикобаламин) 400-500 мкг/сут в/м (30-40 инъекций), затем – поддерживающая доза 500 мкг 1 раз в неделю 3 месяца, затем 500 мкг 1 раз в 2 недели еще 3 мес.
При фуникулярной миелозе 1000 мкг/сут + кобамид (кофермент витамина) 500 мкг/сут в/м до исчезновения признаков поражения спинного мозга.
При фолиеводефицитной анемии: фолиевая кислота внутрь по 10-15 мг/сут до 6 недель.
Гемолитические анемии (ГА)– группа анемий, обусловленных снижением средней продолжительности жизни эритроцитов в результате их усиленного гемолиза (внутриклеточного, тканевого – чаще всего в селезенке или внеклеточного, внутрисосудистого).
В норме продолжительность жизни эритроцитов 100-120 дней, при гемолитической анемии она укорачивается до 12-14 суток.
Признаки ГА: 1) выраженный ретикулоцитоз в периферической крови (признак хорошей регенераторной способности костного мозга); 2) реактивная гиперплазия красного ростка костного мозга; 3) неконьюгированная гипербилирубинемия; 4) гипергемоглобинемия (при внесосудистом гемолизе).
Классификация ГА:
1. По этиологии:
а) наследственные ГА:
1) мембранопатии - наследственный микросфероцитоз (болезнь Минковского-Шоффара)– снижение количества спектрина клеточной мембраны эритроцитов повышает ее проницаемость для воды и ионов натрия; клетки набухают, из дискоцитов превращаются в микросфероциты, которые гибнут в селезенке, а их гемоглобин поглощается макрофагами (внутриклеточный гемолиз); клинически: анемия, желтушность, спленомегалия, гиперхромный кал, могут быть пигментные камни в желчном пузыре, нарушение костеобразования (из-за очагов кроветворения); ОАК: нормохромная анемия, микросфероциты, ретикулоциты; сниженная осмотическая резистентность эритроцитов в гипотонических растворах (в норме 0,36-0,42); миелограмма: соотношение лейкоциты/эритроциты 1:1-1:2 при норме 3:1-4:1; лечение: спленэктомия
2) ферментопатии - дефицит активности глюкозо-6-фосфатдегидрогеназы эритроцитов; клинически анемия, желтушность и другие признаки гемолиза при воздействии агентов, провоцирующих гемолиз (СА, конские бобы, черника, голубика); ОАК – тельца Гейнца (деградация Hb).
3) гемоглобинопатии – нарушения структуры гемоглобина:
а. количественная – талассемия - из-за наличия мутантного гена происходит торможение синтеза цепей глобина; клинически нарушения скелета (квадратный череп), болезнь Кули (желтуха, гепатоспленомегалия, гипохромная микроцитарная анемия, снижение осмотической резистентности эритроцитов – при гомозиготный вариант) или умеренная гипохромная анемия (при гетерозиготном варианте); в ОАК – много овальных и грушевидных эритроцитов
б. качественная – гемоглобинопатия S (серповидно-клеточная анемия) - в HbS в положении 6 четвертого пептида β-цепи гидрофильная глютаминовая кислота заменена на валин, поэтому гемоглобин начинает кристаллизоваться; клинически – анемия, высокий башенный череп, желтушность; часты осложнения – инфаркты органов, гемолитические кризи, тромботические осложнения в любом возрасте; ОАК – деформированные (вытянутые, серповидные) эритроциты
б) приобретенные ГА:
1) при гиперспленизме
2) иммунные ГА: аутоиммунные, лекарственные и др.
3) обусловленные механическим повреждением эритроцитов: маршевая гемоглобинурия, ГА при протезировании клапанов сердца и сосудов
4) токсические: при укусе змей и пауков, интоксикации рядом металлов (медь), органическими растворителями
5) пароксизмальная ночная гемоглобинурия
2. По серологическому принципу – различные типы аутоиммунных гемолитических анемий (АИГА) – возникают при срыве иммунологической толерантности к неизмененным АГ собственных эритроцитов или к АГ, имеющим сходные с эритроцитами детерминанты
1) АИГА с неполными тепловыми агглютининами – наиболее частый вариант; фиксация IgG и IgA на поверхности эритроцитов с последующей гибелью эритроцитов в селезенке (ее макрофаги имеют рецепторы к Fc-фрагменту иммуноглобулина и осуществляют иммунный фагоцитоз)
Клиника: острое, внезапное начало с появлением слабости, одышки, артралгии, болей в пояснице, повышения температуры тела (из-за распада эритроцитов); больные бледные, характерна спленомегалия, пальпаторно селезенка безболезненна; при частых гемолитических кризах – вторичный токсический гемолитический гепатит; кал темного цвета (повышен стеркобилин), моча светло-желтая (уробилин отсутствует)
Диагностика: нормохромная анемия со значительным снижением гемоглобина (при гемолитическом кризе – до 30-40 г/л), постепенным нарастанием ретикулоцитоза; лейкоцитоз до 10-15*109/л, сдвиг влево до миелоцитов (лейкемоидная реакция миелоидного типа на гемолиз); СОЭ – до 30 мм/ч; тромбоциты в норме или снижены; миелограмма – нормобластический тип кроветворения, иногда с чертами мегалобластического; положительная реакция Кумбса - прямая (выявление фиксированных на эритроцитах антител; при малом количестве Ig может дать отрицательный результат) и непрямая (выявление АТ к эритроцитам в сыворотке крови)
Лечение: преднизолон 1 мг/кг/сут ® уровень гемоглобина через 3 дня не стабилизировался ® преднизолон 2 мг/кг/сут с последующим постепенным снижением дозы (при рецидиве – возрат к лечебной дозе) ® гемолиз не купировался за 6 мес ® спленэктомия ® недостаточный эффект ® азатиоприн 50-100 мг/сут до 6 мес., плазмаферез (но не гемосорбция – усиливает гемолиз); по показаниям - гемотрансфузии (используют только отмытые эритроциты).
2) АИГА с тепловыми гемолизинами – внутрисосудистое разрушение эритроцитов под действием АТ-гемолизинов в присутствии комплемента.
Клиника: чаще подострое начало; бледность, желтушность кожных покровов, анемический синдром, нормальные резмеры селезенки; на высоте гемолиза может начаться ДВС-синдром, микротромбоз мезентериальных сосудов, развиться полиорганная недостаточность.
Диагностика: гипергемоглобинемия и гемоглобинурия (моча черного цвета); БАК: повышение ЛДГ (из-за разрушения клеток); отрицательная реакция Кумбса; положительная качественная реакция на аутогемолиз (в чистую пробирку наливают 2-3 мл крови без стабилизатора, ставят ее в термостат на 24 часа при температуре 37 °С, при этом происходит гемолиз, т.к. аутоантитела в сыворотке при t = 37°С фиксируются на поверхности эритроцитов, затем присоединяется комплемент и мембрана эритроцитов разрушается)
Лечение: преднизолон 1-2 мг/кг ® нет эффекта ® цитостатики; при гиперкоагуляции: гепарин, реополиглюкин, при кровоточивости – альбумин, сыворотка, плазмаферез (но не СЗП – приводит к гемолизу из-за содержания в ней комплемента); при гемоглобине ниже 70 г/л – переливание отмытой эритроцитарной массы.
3) АИГА с полными холодовыми агглютининами –фиксация холодовых аутоантител на АГ собственных эритроцитах с их последующей внутрикапиллярной агглютинацией при температуре 4-15 °С и возникновением синдрома повышенной вязкости (гипервискозности).
Клиника: в холодную погоду наблюдается посинение и отек кожи лица, синюшная кожа рук, ног, ушей, синдром Рейно (вплоть до гангрены), летом симптомы проходят; желтуха отсутствует (гипербилирубинемия не характерна, т.к. происходит не гемолиз, а агглютинация); селезенка не увеличена.
Диагностика: умеренное снижение гемоглобина, невысокий ретикулоцитоз, лейкоцитоз до 10-12*109/л, СОЭ увеличена до 70 мм/час и более; осмотическая резистентность эритроцитов не изменена; реакция Кумбса отрицательная.
Лечение: коррекция синдрома гипервискозности: плазмаферез 4-5 процедур (возвращаемые эритроциты подогревают до 37°С) + реополиглюкин как дезагрегант (подогретый 37 °С); при гемотрансфузии – переливание отмытой, подогретой эритроцитарной массы
4) АИГА с двухфазными гемолизинами Доната-Ландштейнера – фиксация IgG на эритроцитах с участием комплемента при низких температурах с последующим их внутрисосудистым гемолизом после согревания
Клиника: вскоре после переохлаждения появляется головная боль, боли в ногах и поясничной области, чувство «ломоты в теле», озноб, повышается температура тела, рвота, черная моча (гемолитический криз);после криза характерны желтушность кожи, увеличение печени и селезенки, иногда – синдром Рейно и холодовая крапивница.
Диагностика: во время криза – нормохромная анемия с ретикулоцитозом, СОЭ умеренно увеличена, лейкоцитоз; гемоглобинурия; серологический анализ крови: если пробирку с кровью поставить на час в холодильник, а затем согреть в термостате до температуры 37 °С, - происходит гемолиз.
Лечение: ГКС в дозах 1-2 мг/кг, при неэффективности – цитостатики
Апластические анемии (АА) – группа патологических состояний, характеризующихся панцитопенией, уменьшением клеточности (абсолютного содержания клеток) костного мозга при содержании бластов менее 5%.
Классификация апластических анемий по этиологии:
а) врожденная (анемия Фанкони, анемия Джозефа-Дайемонда-Блекфена и др.)
б) приобретенная вследствие:
1) воздействия химических веществ (бензол, пестициды, соединения свинца)
2) воздействия лекарственных средств (хлорамфеникол, фенитоин, фенилбутазон, соединения золота и др.)
3) аутоагрессии и появления аутоантител к кроветворным клеткам
4) воздействия радиоактивного излучения
5) инфекционных заболеваний (ВГВ, ВГС, инфекционный мононуклеоз, цитомегаловирусная инфекция)
6) других заболеваний (тимомы, лимфомы и др.)
в) идиопатическая (с неустановленной причиной)
Патогенез апластических анемий: генетические нарушения, воздействие агрессивных веществ, аутоиммунные реакции ® нарушение созревания частично детерминированной стволовой клетки ® уменьшение количества стволовых клеток ® уменьшение количества эритроцитов, тромбоцитов, лейкоцитов в периферической крови.
Клиника апластических анемий:
а) приобретенная апластическая анемия: постепенное или острое начало с быстро нарастающей панцитопенией, выраженным анемическим синдромом, тяжелым геморрагическим синдромом и инфекционными осложнениями; иногда заболевание осложняется внутрисосудистым гемолизом (появляются желтушность кожи и видимых слизистых, темная моча, незначительное увеличение печени, тромбозы множественной локализации).
б) анемия Фанкони: чаще начинается в 5-10 лет; вместе с нарушениями гемопоэза характерны низкий рост, врожденные дефекты скелета (микроцефалия, аномалии фаланг пальцев), аномалии мочеполовой системы (подковообразная почка и др.), гипер- или гипопигментация кожи, умственная отсталость
Диагностика апластических анемий:
1. ОАК: панцитопения (эритро-, тромбо-, лейкоцитопения с нейтропенией и относительным лимфоцитозом)
2. Миелограмма: уменьшение количества миелокариоцитов, относительное повышение содержания лимфоцитов, абсолютное снижение количества нейтрофилов, элементов эритропоэза, резкое уменьшение мегакариоцитопоэза
3. Трепанобиопсия с иммунофенотипированием клеток (необходима для установки генеза апластической анемии): жировой костный мозг преобладает над деятельным, очаги кроветворения редки и малоклеточны
Лечение апластических анемий:
1. Госпитализация в гематологический стационар; при резкой нейтропении – изоляция больного в асептический бокс
2. Трансплантация стволовых клеток периферической крови или костного мозга – рекомендуется всем больным до 20 лет, даже при тяжелом течении апластической анемии, особенно показана зависимым от гемотрансфузий больным (у которых нужны переливания крови чаще 1 раза в мес); до трансплантации костного мозга проводят химиотерапию с тотальным облучением тела.
3. Специфическая консервативная терапия – основной метод лечения для больных старше 20 лет при легком течении заболевания или невозможности выполнить трансплантацию костного мозга: антитимоцитарный иммуноглобулин 20 мг/кг/сут в/в капельно 5 дней в асептических условиях; циклоспорин 5 мг/кг/сут внутрь; метилпреднизолон по 2 мг/кг/сут в/в с 1-ого по 14-ый день, затем по 1 мг/кг/сут с 15-ого по 21-ый день; колониестимулирующие факторы - молграмостим 5 мкг/кг/сут п/к до увеличения числа гранулоцитов > 1*109/л.
4. Спленэктомия (наиболее эффективна при нетяжелой апластической анемии)
5. Профилактика и лечение осложнений (анемического синдрома, геморрагического синдрома, инфекционных осложнений, гемосидероза, сердечной и почечной недостаточности и др.)
Дата добавления: 2015-02-10; просмотров: 1678; Мы поможем в написании вашей работы!; Нарушение авторских прав |