КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Влияние температуры на положение равновесия
Повышение либо понижение температуры означает приобретение либо потерю системой энергии и, следовательно, должно изменять величину константы равновесия.
Запишем уравнение (I.99) в следующем виде:
 (I.104)
(I.104)
 (I.105)
(I.105)
Продифференцировав выражение (I.105) по температуре, получаем для зависимости константы равновесия от температуры уравнение (I.106) – изобару Вант-Гоффа:
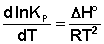 (I.06)
(I.06)
Рассуждая аналогичным образом, для процесса, проходящего в изохорных условиях, можно получить изохору Вант-Гоффа:
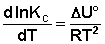 (I.107)
(I.107)
Изобара и изохора Вант-Гоффа связывают изменение константы химического равновесия с тепловым эффектом реакции в изобарных и изохорных условиях соответственно. Очевидно, что чем больше по абсолютной величине тепловой эффект химической реакции, тем сильнее влияет температура на величину константы равновесия. Если реакция не сопровождается тепловым эффектом, то константа равновесия не зависит от температуры.
Экзотермические реакции: ΔH° < 0 (ΔU° < 0). В этом случае, согласно (I.106, I.107), температурный коэффициент логарифма константы равновесия отрицателен. Повышение температуры уменьшает величину константы равновесия, т.е. смещает равновесие влево.
Эндотермические реакции: ΔH° > 0 (ΔU° > 0). В этом случае температурный коэффициент логарифма константы равновесия положителен; повышение температуры увеличивает величину константы равновесия (смещает равновесие вправо).
Графики зависимостей константы равновесия от температуры для экзотермических и эндотермических реакций приведены на рис. I.4.

Рис. 1.4Зависимость константы равновесия от температуры.
Действие рассмотренных нами факторов (давления, концентрации и температуры), равно как и любых других, на систему, находящуюся в состоянии равновесия, обобщает принцип смещения равновесия, называемый также принципом Ле Шателье – Брауна:
Если на систему, находящуюся в состоянии истинного равновесия, оказывается внешнее воздействие, то в системе возникает самопроизвольный процесс, компенсирующий данное воздействие.
Принцип Ле Шателье – Брауна является одним из следствий второго начала термодинамики и применим к любым макроскопическим системам, находящимся в состоянии истинного равновесия.
1.8 ФАЗОВЫЕ РАВНОВЕСИЯ
Вещество при изменении давления и температуры может переходить из одного агрегатного состояния в другое. Эти переходы, совершающиеся при постоянной температуре, называют фазовыми переходами первого рода. Количество теплоты, которое вещество получает из окружающей среды либо отдает окружающей среде при фазовом переходе, есть скрытая теплота фазового перехода λфп. Если рассматривается гетерогенная система, в которой нет химических взаимодействий, а возможны лишь фазовые переходы, то при постоянстве температуры и давления в системе существует т.н. фазовое равновесие. Фазовое равновесие характеризуется некоторым числом фаз, компонентов и числом степеней термодинамической свободы системы.
Компонент – химически однородная составная часть системы, которая может быть выделена из системы и существовать вне её. Число независимых компонентов системы равно числу компонентов минус число возможных химических реакций между ними. Число степеней свободы – число параметров состояния системы, которые могут быть одновременно произвольно изменены в некоторых пределах без изменения числа и природы фаз в системе.
Число степеней свободы гетерогенной термодинамической системы, находящейся в состоянии фазового равновесия, определяется правилом фаз, сформулированным Дж. Гиббсом:
Число степеней свободы равновесной термодинамической системы С равно числу независимых компонентов системы К минус число фаз Ф плюс число внешних факторов, влияющих на равновесие.
Для системы, на которую из внешних факторов влияют только температура и давление, можно записать:
С = К – Ф + 2 (I.108)
Системы принято классифицировать по числу компонентов (одно-, двухкомпонентные и т.д.), по числу фаз (одно-, двухфазные и т.д.) и числу степеней свободы (инвариантные, моно-, дивариантные и т.д.). Для систем с фазовыми переходами обычно рассматривают графическую зависимость состояния системы от внешних условий – т.н. диаграммы состояния.
Анализ диаграмм состояния позволяет определить число фаз в системе, границы их существования, характер взаимодействия компонентов. В основе анализа диаграмм состояния лежат два принципа: принцип непрерывности и принцип соответствия. Согласно принципу непрерывности, при непрерывном изменении параметров состояния все свойства отдельных фаз изменяются также непрерывно; свойства системы в целом изменяются непрерывно до тех пор, пока не изменится число или природа фаз в системе, что приводит к скачкообразному изменению свойств системы. Согласно принципу соответствия, на диаграмме состояния системы каждой фазе соответствует часть плоскости – поле фазы. Линии пересечения плоскостей отвечают равновесию между двумя фазами. Всякая точка на диаграмме состояния (т. н. фигуративная точка) отвечает некоторому состоянию системы с определенными значениями параметров состояния.
Рассмотрим и проанализируем диаграмму состояния воды (рис.1.4). Поскольку вода – единственное присутствующее в системе вещество, число независимых компонентов К = 1. В системе возможны три фазовых равновесия: между жидкостью и газом (линия ОА – зависимость давления насыщенного пара воды от температуры), твердым телом и газом (линия ОВ – зависимость давления насыщенного пара надо льдом от температуры), твердым телом и жидкостью (линия ОС – зависимость температуры плавления льда от давления). Три кривые имеют точку пересечения О, называемую тройной точкой воды; тройная точка отвечает равновесию между тремя фазами.
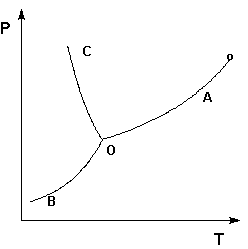
Рис. 1.4. Диаграмма состояния воды
В тройной точке система трехфазна и число степеней свободы равно нулю; три фазы могут находиться в равновесии лишь при строго определенных значениях температуры и давления (для воды тройная точка отвечает состоянию с Р = 6.1 кПа и Т = 273.16 К).
Кривая ОВ теоретически продолжается до абсолютного нуля, а кривая давления насыщенного пара над жидкостью ОА заканчивается в критической точке воды (Tкр = 607.46 К, Ркр = 19.5 МПа); выше критической температуры газ и жидкость не могут существовать как отдельные фазы. Кривая ОС в верхней части (при высоких давлениях) изменяет свой наклон (появляются новые кристаллические фазы, плотность которых, в отличие от обычного льда, выше, чем у воды).
Внутри каждой из областей диаграммы (АОВ, ВОС, АОС) система однофазна; число степеней свободы системы равно двум (система дивариантна), т.е. можно одновременно изменять и температуру, и давление, не вызывая изменения числа фаз в системе:
С = 1 – 1 + 2 = 2
На каждой из линий число фаз в системе равно двум и, согласно правилу фаз, система моновариантна, т.е. для каждого значения температуры имеется только одно значение давления, при котором система двухфазна:
С = 1 – 2 + 2 = 1
Влияние давления на температуру фазового перехода описывает уравнение Клаузиуса – Клапейрона:
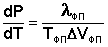 (I.109)
(I.109)
Здесь ΔVфп = V2 – V1 есть изменение молярного объема вещества при фазовом переходе (причем V2 относится к состоянию, переход в которое сопровождается поглощением теплоты). Уравнение Клаузиуса – Клапейрона позволяет объяснить наклон кривых равновесия на диаграмме состояния однокомпонентной системы. Для переходов "жидкость – пар" и "твердое вещество – пар" ΔV всегда больше нуля; поэтому кривые на диаграмме состояния, отвечающие этим равновесиям, всегда наклонены вправо (повышение температуры всегда увеличивает давление насыщенного пара). Поскольку молярный объем газа много больше молярного объема того же вещества в жидком или твердом агрегатном состояниях (Vг >> Vж, Vг >> Vт), уравнение (I.109) для частных случаев испарения и возгонки примет следующий вид:
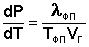 (I.110)
(I.110)
Для многих веществ скрытая теплота парообразования или возгонки постоянна в большом интервале температур; в этом случае уравнение (I.110) можно проинтегрировать:
 (I.111)
(I.111)
Кривая равновесия "твердое вещество – жидкость" на диаграммах состояния воды и висмута наклонена влево, а на диаграммах состояния остальных веществ – вправо. Это связано с тем, что плотность воды больше, чем плотность льда (и плотность жидкого висмута больше его плотности в твердом состоянии), т.е. плавление сопровождается уменьшением объема (ΔV < 0). Как следует из выражения (I.111), в этом случае увеличение давления будет понижать температуру фазового перехода "твердое тело – жидкость" (воду и висмут относят поэтому к т.н. аномальным веществам). Для всех остальных веществ (т.н. нормальные вещества) ΔVпл > 0 и, согласно уравнению Клаузиуса – Клапейрона, увеличение давления приводит к повышению температуры плавления.
2 ХИМИЧЕСКАЯ КИНЕТИКА
Законы химической термодинамики позволяют определить направление и предел протекания возможного при данных условиях химического процесса, а также его энергетический эффект. Однако термодинамика не может ответить на вопросы о том, как осуществляется данный процесс и с какой скоростью. Эти вопросы – механизм и скорость химической реакции – и являются предметом химической кинетики.
2.1 Скорость химической реакции
Дадим определение основному понятию химической кинетики – скорости химической реакции:
Скорость химической реакции есть число элементарных актов химической реакции, происходящих в единицу времени в единице объема (для гомогенных реакций) или на единице поверхности (для гетерогенных реакций).
Скорость химической реакции есть изменение концентрации реагирующих веществ в единицу времени.
Первое определение является наиболее строгим; из него следует, что скорость химической реакции можно также выражать как изменение во времени любого параметра состояния системы, зависящего от числа частиц какого-либо реагирующего вещества, отнесенное к единице объема или поверхности – электропроводности, оптической плотности, диэлектрической проницаемости и т.д. и т.п. Однако наиболее часто в химии рассматривается зависимость концентрации реагентов от времени. В случае односторонних (необратимых) химических реакций (здесь и далее рассматриваются только односторонние реакции) очевидно, что концентрации исходных веществ во времени постоянно уменьшаются (ΔСисх < 0), а концентрации продуктов реакции увеличиваются (ΔСпрод > 0). Скорость реакции считается положительной, поэтому математически определение средней скорости реакции в интервале времени Δt записывается следующим образом:
 (II.1)
(II.1)
В различных интервалах времени средняя скорость химической реакции имеет разные значения; истинная (мгновенная) скорость реакции определяется как производная от концентрации по времени:
 (II.2)
(II.2)
Графическое изображение зависимости концентрации реагентов от времени есть кинетическая кривая (рисунок 2.1).

Рис. 2.1Кинетические кривые для исходных веществ (А) и продуктов реакции (В).
Истинную скорость реакции можно определить графически, проведя касательную к кинетической кривой (рис. 2.2); истинная скорость реакции в данный момент времени равна по абсолютной величине тангенсу угла наклона касательной:

Рис. 2.2 Графическое определение Vист.
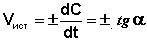 (II.3)
(II.3)
Необходимо отметить, что в том случае, если стехиометрические коэффициенты в уравнении химической реакции неодинаковы, величина скорости реакции будет зависеть от того, изменение концентрации какого реагента определялось. Очевидно, что в реакции
2Н2 + О2 ––> 2Н2О
концентрации водорода, кислорода и воды изменяются в различной степени:
ΔС(Н2) = ΔС(Н2О) = 2 ΔС(О2).
Скорость химической реакции зависит от множества факторов: природы реагирующих веществ, их концентрации, температуры, природы растворителя и т.д.
2.1.1 Кинетическое уравнение химической реакции. Порядок реакции.
Одной из задач, стоящих перед химической кинетикой, является определение состава реакционной смеси (т.е. концентраций всех реагентов) в любой момент времени, для чего необходимо знать зависимость скорости реакции от концентраций. В общем случае, чем больше концентрации реагирующих веществ, тем больше скорость химической реакции. В основе химической кинетики лежит т. н. основной постулат химической кинетики:
Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в некоторых степенях.
Т. е. для реакции
аА + bВ + dD + ... ––> еЕ + ...
можно записать:
 (II.4)
(II.4)
Коэффициент пропорциональности k есть константа скорости химической реакции. Константа скорости численно равна скорости реакции при концентрациях всех реагирующих веществ, равных 1 моль/л.
Зависимость скорости реакции от концентраций реагирующих веществ определяется экспериментально и называется кинетическим уравнением химической реакции. Очевидно, что для того, чтобы записать кинетическое уравнение, необходимо экспериментально определить величину константы скорости и показателей степени при концентрациях реагирующих веществ. Показатель степени при концентрации каждого из реагирующих веществ в кинетическом уравнении химической реакции (в уравнении (II.4) соответственно x, y и z) есть частный порядок реакции по данному компоненту. Сумма показателей степени в кинетическом уравнении химической реакции (x + y + z) представляет собой общий порядок реакции. Следует подчеркнуть, что порядок реакции определяется только из экспериментальных данных и не связан со стехиометрическими коэффициентами при реагентах в уравнении реакции. Стехиометрическое уравнение реакции представляет собой уравнение материального баланса и никоим образом не может определять характера протекания этой реакции во времени.
В химической кинетике принято классифицировать реакции по величине общего порядка реакции. Рассмотрим зависимость концентрации реагирующих веществ от времени для необратимых (односторонних) реакций нулевого, первого и второго порядков.
2.1.2 Реакции нулевого порядка
Для реакций нулевого порядка кинетическое уравнение имеет следующий вид:
 (II.5)
(II.5)
Скорость реакции нулевого порядка постоянна во времени и не зависит от концентраций реагирующих веществ; это характерно для многих гетерогенных (идущих на поверхности раздела фаз) реакций в том случае, когда скорость диффузии реагентов к поверхности меньше скорости их химического превращения.
2.1.3 Реакции первого порядка
Рассмотрим зависимость от времени концентрации исходного вещества А для случая реакции первого порядка А ––> В. Реакции первого порядка характеризуются кинетическим уравнением вида (II.6). Подставим в него выражение (II.2):
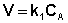 (II.6)
(II.6)
 (II.7)
(II.7)
После интегрирования выражения (II.7) получаем:
 (II.8)
(II.8)
Константу интегрирования g определим из начальных условий: в момент времени t = 0 концентрация С равна начальной концентрации Со. Отсюда следует, что g = ln Со. Получаем:
 (II.9)
(II.9)

Рис. 2.3 Зависимость логарифма концентрации от времени для реакций первого порядка
Т.о., логарифм концентрации для реакции первого порядка линейно зависит от времени (рис. 2.3) и константа скорости численно равна тангенсу угла наклона прямой к оси времени.
 (II.10)
(II.10)
Из уравнения (II.9) легко получить выражение для константы скорости односторонней реакции первого порядка:
 (II.11)
(II.11)
Еще одной кинетической характеристикой реакции является период полупревращения t1/2 – время, за которое концентрация исходного вещества уменьшается вдвое по сравнению с исходной. Выразим t1/2 для реакции первого порядка, учитывая, что С = ½Со:
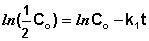 (II.12)
(II.12)
Отсюда
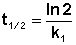 (II.13)
(II.13)
Как видно из полученного выражения, период полупревращения реакции первого порядка не зависит от начальной концентрации исходного вещества.
2.1.4 Реакции второго порядка
Для реакций второго порядка кинетическое уравнение имеет следующий вид:
 (II.14)
(II.14)
либо
 (II.15)
(II.15)
Рассмотрим простейший случай, когда кинетическое уравнение имеет вид (II.14) или, что то же самое, в уравнении вида (II.15) концентрации исходных веществ одинаковы; уравнение (II.14) в этом случае можно переписать следующим образом:
 (II.16)
(II.16)
После разделения переменных и интегрирования получаем:
 (II.17)
(II.17)
Постоянную интегрирования g, как и в предыдущем случае, определим из начальных условий. Получим:
 (II.18)
(II.18)
Т.о., для реакций второго порядка, имеющих кинетическое уравнение вида (II.14), характерна линейная зависимость обратной концентрации от времени (рис. 2.4) и константа скорости равна тангенсу угла наклона прямой к оси времени:
 (II.19)
(II.19)
 (II.20)
(II.20)

Рис. 2.4 Зависимость обратной концентрации от времени для реакций второго порядка
Если начальные концентрации реагирующих веществ Cо,А и Cо,В различны, то константу скорости реакции находят интегрированием уравнения (II.21), в котором CА и CВ – концентрации реагирующих веществ в момент времени t от начала реакции:
 (II.21)
(II.21)
В этом случае для константы скорости получаем выражение
 (II.22)
(II.22)
Порядок химической реакции есть формально-кинетическое понятие, физический смысл которого для элементарных (одностадийных) реакций заключается в следующем: порядок реакции равен числу одновременно изменяющихся концентраций. В случае элементарных реакций порядок реакции может быть равен сумме коэффициентов в стехиометрическом уравнении реакции; однако в общем случае порядок реакции определяется только из экспериментальных данных и зависит от условий проведения реакции. Рассмотрим в качестве примера элементарную реакцию гидролиза этилового эфира уксусной кислоты (этилацетата), кинетика которой изучается в лабораторном практикуме по физической химии:
СН3СООС2Н5 + Н2О ––> СН3СООН + С2Н5ОН
Если проводить эту реакцию при близких концентрациях этилацетата и воды, то общий порядок реакции равен двум и кинетическое уравнение имеет следующий вид:
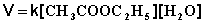 (II.23)
(II.23)
При проведении этой же реакции в условиях большого избытка одного из реагентов (воды или этилацетата) концентрация вещества, находящегося в избытке, практически не изменяется и может быть включена в константу скорости; кинетическое уравнение для двух возможных случаев принимает следующий вид:
1) Избыток воды:
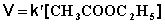 (II.24)
(II.24)
 (II.25)
(II.25)
2) Избыток этилацетата:
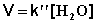 (II.26)
(II.26)
 (II.27)
(II.27)
В этих случаях мы имеем дело с т.н. реакцией псевдопервого порядка. Проведение реакции при большом избытке одного из исходных веществ используется для определения частных порядков реакции.
2.1.5 Методы определения порядка реакции
Проведение реакции в условиях, когда концентрация одного из реагентов много меньше концентрации другого (других) и скорость реакции зависит от концентрации только этого реагента, используется для определения частных порядков реакции – это т.н. метод избыточных концентраций или метод изолирования Оствальда. Порядок реакции по данному веществу определяется одним из перечисленных ниже методов.
Графический метод заключается в построении графика зависимости концентрации реагента от времени в различных координатах. Для различных частных порядков эти зависимости имеют следующий вид:
| Порядок реакции | Зависимость концентрации от времени |

| |

| |

|
Если построить графики этих зависимостей на основании опытных данных, то лишь одна из них будет являться прямой линией. Если, например, график, построенный по опытным данным, оказался прямолинейным к координатах lnC = f(t), то частный порядок реакции по данному веществу равен единице.
Метод подбора кинетического уравнения заключается в подстановке экспериментальных данных изучения зависимости концентрации вещества от времени в кинетические уравнения различных порядков. Подставляя в приведённые в таблице уравнения значения концентрации реагента в разные моменты времени, вычисляют значения константы скорости. Частный порядок реакции по данному веществу равен порядку того кинетического уравнения, для которого величина константы скорости остаётся постоянной во времени.
| Порядок реакции | Выражение для константы скорости |

| |
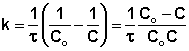
| |

|
Метод определения времени полупревращения заключается в определении t1/2 для нескольких начальных концентраций. Как видно из приведённых в таблице уравнений, для реакции первого порядка время полупревращения не зависит от Co, для реакции второго порядка – обратно пропорционально Co, и для реакции третьего порядка – обратно пропорционально квадрату начальной концентрации.
| Порядок реакции | Выражение для периода полупревращения |
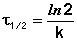
| |

| |

|
По характеру зависимости t1/2 от Co нетрудно сделать вывод о порядке реакции по данному веществу. Данный метод, в отличие от описанных выше, применим и для определения дробных порядков.
2.1.6 Молекулярность элементарных реакций
Элементарными (простыми) называют реакции, идущие в одну стадию. Их принято классифицировать по молекулярности:
Молекулярность элементарной реакции – число частиц, которые, согласно экспериментально установленному механизму реакции, участвуют в элементарном акте химического взаимодействия.
Мономолекулярные – реакции, в которых происходит химическое превращение одной молекулы (изомеризация, диссоциация и т. д.):
I2 ––> I• + I•
Бимолекулярные – реакции, элементарный акт которых осуществляется при столкновении двух частиц (одинаковых или различных):
СН3Вr + КОН ––> СН3ОН + КВr
Тримолекулярные – реакции, элементарный акт которых осуществляется при столкновении трех частиц:
О2 + NО + NО ––> 2NО2
Реакции с молекулярностью более трёх неизвестны.
Для элементарных реакций, проводимых при близких концентрациях исходных веществ, величины молекулярности и порядка реакции совпадают. Тем не менее, никакой чётко определенной взаимосвязи между понятиями молекулярности и порядка реакции не существует, поскольку порядок реакции характеризует кинетическое уравнение реакции, а молекулярность – механизм реакции.
2.1.7 Сложные реакции
Сложными называют химические реакции, протекающие более чем в одну стадию. Рассмотрим в качестве примера одну из сложных реакций, кинетика и механизм которой хорошо изучены:
2НI + Н2О2 ––> I2 + 2Н2О
Данная реакция является реакцией второго порядка; её кинетическое уравнение имеет следующий вид:
 (II.28)
(II.28)
Изучение механизма реакции показало, что она является двухстадийной (протекает в две стадии):
1) НI + Н2О2 ––> НIО + Н2О
2) НIО + НI ––> I2 + Н2О
Скорость первой стадии V1 много больше скорости второй стадии V2 и общая скорость реакции определяется скоростью более медленной стадии, называемой поэтому скоростьопределяющей или лимитирующей.
Сделать вывод о том, является реакция элементарной или сложной, можно на основании результатов изучения её кинетики. Реакция является сложной, если экспериментально определенные частные порядки реакции не совпадают с коэффициентами при исходных веществах в стехиометрическом уравнении реакции; частные порядки сложной реакции могут быть дробными либо отрицательными, в кинетическое уравнение сложной реакции могут входить концентрации не только исходных веществ, но и продуктов реакции.
2.1.8 Классификация сложных реакций
Дата добавления: 2014-12-03; просмотров: 377; Мы поможем в написании вашей работы!; Нарушение авторских прав |