КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
А ––> K# ––> B
Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е'А выше, нежели энергия активации прямой реакции EA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции (рис. 2.5).
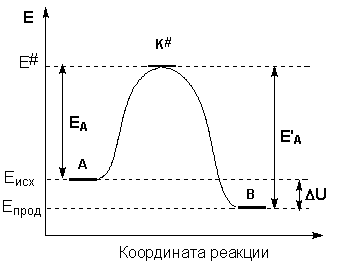
Рис. 2.5 Энергетическая диаграмма химической реакции.
Eисх – средняя энергия частиц исходных веществ,
Eпрод – средняя энергия частиц продуктов реакции
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции (рис.2.6):

Рис. 2.6 Распределение частиц по энергии Здесь nЕ/N – доля частиц, обладающих энергией E; Ei - средняя энергия частиц при температуре Ti (T1 < T2 < T3)
Рассмотрим термодинамический вывод выражения, описывающего зависимость константы скорости реакции от температуры и величины энергии активации – уравнения Аррениуса. Согласно уравнению изобары Вант-Гоффа,
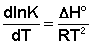 (II.31)
(II.31)
Поскольку константа равновесия есть отношение констант скоростей прямой и обратной реакции, можно переписать выражение (II.31) следующим образом:
 (II.32)
(II.32)
Представив изменение энтальпии реакции ΔHº в виде разности двух величин E1 и E2, получаем:
 (II.33)
(II.33)
 (II.34)
(II.34)
Здесь С – некоторая константа. Постулировав, что С = 0, получаем уравнение Аррениуса, где EA – энергия активации:
 (II.35)
(II.35)
После неопределенного интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
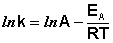 (II.36)
(II.36)
 (II.37)
(II.37)

Рис. 2.7 Зависимость логарифма константы скорости химической реакции от обратной температуры.
Здесь A – постоянная интегрирования. Из уравнения (II.37) нетрудно показать физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения (II.36), логарифм константы скорости линейно зависит от обратной температуры (рис.2.7); величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
 (II.38)
(II.38)
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
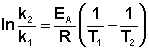 (II.39)
(II.39)
2.1.11 Кинетика двусторонних (обратимых) реакций
Химические реакции часто являются двусторонними (обратимыми), т.е. могут протекать при данных условиях в двух противоположных направлениях (понятие обратимая реакция следует отличать от термодинамического понятия обратимый процесс; двусторонняя реакция обратима в термодинамическом смысле лишь в состоянии химического равновесия). Рассмотрим элементарную двустороннюю реакцию
А + В <––> D + E
Скорость уменьшения концентрации вещества А при протекании прямой реакции определяется уравнением (II.40):
 , (II.40)
, (II.40)
а скорость возрастания концентрации вещества А в результате протекания обратной реакции – уравнением (II.41):
 (II.41)
(II.41)
Общая скорость двусторонней реакции в любой момент времени равна разности скоростей прямой и обратной реакции:
 (II.42)
(II.42)
По мере протекания двусторонней реакции скорость прямой реакции уменьшается, скорость обратной реакции – увеличивается; в некоторый момент времени скорости прямой и обратной реакции становятся равными и концентрации реагентов перестают изменяться. Таким образом, в результате протекания в закрытой системе двусторонней реакции система достигает состояния химического равновесия; при этом константа равновесия будет равна отношению констант скоростей прямой и обратной реакции:
 (II.43)
(II.43)
2.1.12 Кинетика гетерогенных химических реакций
Когда реакция совершается между веществами, находящимися в разных фазах гетерогенной системы, основной постулат химической кинетики становится неприменимым. В гетерогенных реакциях роль промежуточных продуктов обычно играют молекулы, связанные химическими силами с поверхностью раздела фаз (химически адсорбированные на поверхности). Во всяком гетерогенном химическом процессе можно выделить следующие стадии:
1. Диффузия реагентов к реакционной зоне, находящейся на поверхности раздела фаз.
2. Активированная адсорбция частиц реагентов на поверхности.
3. Химическое превращение адсорбированных частиц.
4. Десорбция образовавшихся продуктов реакции.
5. Диффузия продуктов реакции из реакционной зоны.
Стадии 1 и 5 называются диффузионными, стадии 2, 3 и 4 – кинетическими. Универсального выражения для скорости гетерогенных химических реакций не существует, поскольку каждая из выделенных стадий может являться лимитирующей. Как правило, при низких температурах скорость гетерогенной реакции определяют кинетические стадии (т.н. кинетическая область гетерогенного процесса; скорость реакции в этом случае сильно зависит от температуры и величины площади поверхности раздела фаз; порядок реакции при этом может быть любым). При высоких температурах скорость процесса будет определяться скоростью диффузии (диффузионная область гетерогенной реакции, характеризующаяся, как правило, первым порядком реакции и слабой зависимостью скорости процесса от температуры и площади поверхности раздела фаз).
2.2 ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Передача энергии для активации вступающих во взаимодействие молекул может осуществляться либо в форме теплоты (т. н. темновые реакции), либо в виде квантов электромагнитного излучения. Реакции, в которых активация частиц является результатом их взаимодействия с квантами электромагнитного излучения видимой области спектра, называют фотохимическими реакциями. При всех фотохимических процессах выполняется закон Гротгуса:
Химическое превращение вещества может вызвать только то излучение, которое поглощается этим веществом.
Излучение, отражённое веществом, а также прошедшее сквозь него, не вызывают никаких химических превращений. Иногда фотохимические процессы происходят под действием излучения, которое не поглощается реагирующими веществами; однако в таких случаях реакционная смесь должна содержать т.н. сенсибилизаторы. Механизм действия сенсибилизаторов заключается в том, что они поглощают свет, переходя в возбуждённое состояние, а затем при столкновении с молекулами реагентов передают им избыток своей энергии. Сенсибилизатором фотохимических реакций является, например, хлорофилл (см. ниже).
Взаимодействие света с веществом может идти по трём возможным направлениям:
1. Возбуждение частиц (переход электронов на вышележащие орбитали):
A + hν ––> A*
2. Ионизация частиц за счет отрыва электронов:
A + hν ––> A+ + e–
3. Диссоциация молекул с образованием свободных радикалов (гомолитическая) либо ионов (гетеролитическая):
AB + hν ––> A• + B•
AB + hν ––> A+ + B–
Между количеством лучистой энергии, поглощенной молекулами вещества, и количеством фотохимически прореагировавших молекул существует соотношение, выражаемое законом фотохимической эквивалентности Штарка – Эйнштейна:
Число молекул, подвергшихся первичному фотохимическому превращению, равно числу поглощенных веществом квантов электромагнитного излучения.
Поскольку фотохимическая реакция, как правило, включает в себя и т.н. вторичные процессы (например, в случае цепного механизма), для описания реакции вводится понятие квантовый выход фотохимической реакции:
Квантовый выход фотохимической реакции γ есть отношение числа частиц, претерпевших превращение, к числу поглощенных веществом квантов света.
Квантовый выход реакции может варьироваться в очень широких пределах: от 10-3 (фотохимическое разложение метилбромида) до 106 (цепная реакция водорода с хлором); в общем случае, чем более долгоживущей является активная частица, тем с большим квантовым выходом протекает фотохимическая реакция.
Важнейшими фотохимическими реакциями являются реакции фотосинтеза, протекающие в растениях с участием хлорофилла:

Процесс фотосинтеза составляют две стадии: световая, связанная с поглощением фотонов, и значительно более медленная темновая, представляющая собой ряд химических превращений, осуществляемых в отсутствие света. Суммарный процесс фотосинтеза заключается в окислении воды до кислорода и восстановлении диоксида углерода до углеводов:
СО2 + Н2О + hν ––> (СН2О) + О2, ΔG° = 477.0 кДж/моль
Протекание данного окислительно-восстановительного процесса (связанного с переносом электронов) возможно благодаря наличию в реакционном центре хлорофилла Сhl донора D и акцептора A электронов; перенос электронов происходит в результате фотовозбуждения молекулы хлорофилла:
DChlA + hν ––> DChl*A ––> DChl+A– ––> D+ChlA–
Возникающие в данном процессе заряженные частицы D+ и A– принимают участие в дальнейших окислительно-восстановительных реакциях темновой стадии фотосинтеза.
2.3 КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ
Скорость химической реакции при данной температуре определяется скоростью образования активированного комплекса, которая, в свою очередь, зависит от величины энергии активации. Во многих химических реакциях в структуру активированного комплекса могут входить вещества, стехиометрически не являющиеся реагентами; очевидно, что в этом случае изменяется и величина энергии активации процесса. В случае наличия нескольких переходных состояний реакция будет идти в основном по пути с наименьшим активационным барьером.
Катализ – явление изменения скорости химической реакции в присутствии веществ, состояние и количество которых после реакции остаются неизменными.
Различают положительный и отрицательный катализ (соответственно увеличение и уменьшение скорости реакции), хотя часто под термином "катализ" подразумевают только положительный катализ; отрицательный катализ называют ингибированием.
Вещество, входящее в структуру активированного комплекса, но стехиометрически не являющееся реагентом, называется катализатором. Для всех катализаторов характерны такие общие свойства, как специфичность и селективность действия.
Специфичность катализатора заключается в его способности ускорять только одну реакцию или группу однотипных реакций и не влиять на скорость других реакций. Так, например, многие переходные металлы (платина, медь, никель, железо и т.д.) являются катализаторами для процессов гидрирования; оксид алюминия катализирует реакции гидратации и т.д.
Селективность катализатора – способность ускорять одну из возможных при данных условиях параллельных реакций. Благодаря этому можно, применяя различные катализаторы, из одних и тех же исходных веществ получать различные продукты:
| [Cu]:СО + Н2 ––> СН3ОН | [Al2О3]: С2Н5ОН ––> С2Н4 + Н2О |
| [Ni]: СО + Н2 ––> СН4 + Н2О | [Cu]: С2Н5ОН ––> СН3СНО + Н2 |
Причиной увеличения скорости реакции при положительном катализе является уменьшение энергии активации при протекании реакции через активированный комплекс с участием катализатора (рис. 2.8).
Поскольку, согласно уравнению Аррениуса, константа скорости химической реакции находится в экспоненциальной зависимости от величины энергии активации, уменьшение последней вызывает значительное увеличение константы скорости. Действительно, если предположить, что предэкспоненциальные множители в уравнении Аррениуса (II.32) для каталитической и некаталитической реакций близки, то для отношения констант скорости можно записать:
 (II.44)
(II.44)
Если ΔEA = –50 кДж/моль, то отношение констант скоростей составит 2,7·106 раз (действительно, на практике такое уменьшение EA увеличивает скорость реакции приблизительно в 105 раз).
Необходимо отметить, что наличие катализатора не влияет на величину изменения термодинамического потенциала в результате процесса и, следовательно, никакой катализатор не может сделать возможным самопроизвольное протекание термодинамически невозможного процесса (процесса, ΔG (ΔF) которого больше нуля). Катализатор не изменяет величину константы равновесия для обратимых реакций; влияние катализатора в этом случае заключается только в ускорении достижения равновесного состояния.
В зависимости от фазового состояния реагентов и катализатора различают гомогенный и гетерогенный катализ.

Рис. 2.8 Энергетическая диаграмма химической реакции без катализатора (1) и в присутствии катализатора (2).
2.3.1 Гомогенный катализ.
Гомогенный катализ – каталитические реакции, в которых реагенты и катализатор находятся в одной фазе. В случае гомогенно-каталитических процессов катализатор образует с реагентами промежуточные реакционноспособные продукты. Рассмотрим некоторую реакцию
А + В ––> С
В присутствии катализатора осуществляются две быстро протекающие стадии, в результате которых образуются частицы промежуточного соединения АК и затем (через активированный комплекс АВК#) конечный продукт реакции с регенерацией катализатора:
А + К ––> АК
АК + В ––> С + К
Примером такого процесса может служить реакция разложения ацетальдегида, энергия активации которой EA = 190 кДж/моль:
СН3СНО ––> СН4 + СО
В присутствии паров йода этот процесс протекает в две стадии:
СН3СНО + I2 ––> СН3I + НI + СО
СН3I + НI ––> СН4 + I2
Уменьшение энергии активации этой реакции в присутствии катализатора составляет 54 кДж/моль; константа скорости реакции при этом увеличивается приблизительно в 105 раз. Наиболее распространенным типом гомогенного катализа является кислотный катализ, при котором в роли катализатора выступают ионы водорода Н+.
2.3.2Автокатализ.
Автокатализ – процесс каталитического ускорения химической реакции одним из её продуктов. В качестве примера можно привести катализируемую ионами водорода реакцию гидролиза сложных эфиров. Образующаяся при гидролизе кислота диссоциирует с образованием протонов, которые ускоряют реакцию гидролиза. Особенность автокаталитической реакции состоит в том, что данная реакция протекает с постоянным возрастанием концентрации катализатора. Поэтому в начальный период реакции скорость её возрастает, а на последующих стадиях в результате убыли концентрации реагентов скорость начинает уменьшаться; кинетическая кривая продукта автокаталитической реакции имеет характерный S-образный вид (рис. 2.9).

Рис. 2.9 Кинетическая кривая продукта автокаталитической реакции
2.3.3 Гетерогенный катализ.
Гетерогенный катализ – каталитические реакции, идущие на поверхности раздела фаз, образуемых катализатором и реагирующими веществами. Механизм гетерогенно-каталитических процессов значительно более сложен, чем в случае гомогенного катализа. В каждой гетерогенно-каталитической реакции можно выделить как минимум шесть стадий:
1. Диффузия исходных веществ к поверхности катализатора.
2. Адсорбция исходных веществ на поверхности с образованием некоторого промежуточного соединения:
А + В + К ––> АВК
3. Активация адсорбированного состояния (необходимая для этого энергия есть истинная энергия активации процесса):
АВК ––> АВК#
4. Распад активированного комплекса с образованием адсорбированных продуктов реакции:
АВК# ––> СDК
5. Десорбция продуктов реакции с поверхности катализатора.
СDК ––> С + D + К
6. Диффузия продуктов реакции от поверхности катализатора.
Специфической особенностью гетерокаталитических процессов является способность катализатора к промотированию и отравлению.
Промотирование – увеличение активности катализатора в присутствии веществ, которые сами не являются катализаторами данного процесса (промоторов). Например, для катализируемой металлическим никелем реакции
СО + Н2 ––> СН4 + Н2О
введение в никелевый катализатор небольшой примеси церия приводит к резкому возрастанию активности катализатора.
Отравление – резкое снижение активности катализатора в присутствии некоторых веществ (т. н. каталитических ядов). Например, для реакции синтеза аммиака (катализатор – губчатое железо), присутствие в реакционной смеси соединений кислорода или серы вызывает резкое снижение активности железного катализатора; в то же время способность катализатора адсорбировать исходные вещества снижается очень незначительно.
Для объяснения этих особенностей гетерогенно-каталитических процессов Г. Тэйлором было высказано следующее предположение: каталитически активной является не вся поверхность катализатора, а лишь некоторые её участки – т.н. активные центры, которыми могут являться различные дефекты кристаллической структуры катализатора (например, выступы либо впадины на поверхности катализатора). В настоящее время нет единой теории гетерогенного катализа. Для металлических катализаторов была разработана теория мультиплетов. Основные положения мультиплетной теории состоят в следующем:
1. Активный центр катализатора представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности катализатора в геометрическом соответствии со строением молекулы, претерпевающей превращение.
2. При адсорбции реагирующих молекул на активном центре образуется мультиплетный комплекс, в результате чего происходит перераспределение связей, приводящее к образованию продуктов реакции.
Теорию мультиплетов называют иногда теорией геометрического подобия активного центра и реагирующих молекул. Для различных реакций число адсорбционных центров (каждый из которых отождествляется с атомом металла) в активном центре различно – 2, 3, 4 и т.д. Подобные активные центры называются соответственно дублет, триплет, квадруплет и т.д. (в общем случае мультиплет, чему и обязана теория своим названием).
Например, согласно теории мультиплетов, дегидрирование предельных одноатомных спиртов происходит на дублете, а дегидрирование циклогексана – на секстете (рис. 2.10 – 2.11); теория мультиплетов позволила связать каталитическую активность металлов с величиной их атомного радиуса.
 Рис. 2.10 Дегидрирование спиртов на дублете
Рис. 2.10 Дегидрирование спиртов на дублете

Рис. 2.11 Дегидрирование циклогексана на секстете
2.3.4 Ферментативный катализ.
Ферментативный катализ – каталитические реакции, протекающие с участием ферментов – биологических катализаторов белковой природы. Ферментативный катализ имеет две характерные особенности:
1. Высокая активность, на несколько порядков превышающая активность неорганических катализаторов, что объясняется очень значительным снижением энергии активации процесса ферментами. Так, константа скорости реакции разложения перекиси водорода, катализируемой ионами Fе2+, составляет 56 с-1; константа скорости этой же реакции, катализируемой ферментом каталазой, равна 3.5·107, т.е. реакция в присутствии фермента протекает в миллион раз быстрее (энергии активации процессов составляют соответственно 42 и 7.1 кДж/моль). Константы скорости гидролиза мочевины в присутствии кислоты и уреазы различаются на тринадцать порядков, составляя 7.4·10-7 и 5·106 с-1 (величина энергии активации составляет соответственно 103 и 28 кДж/моль).
2. Высокая специфичность. Например, амилаза катализирует процесс расщепления крахмала, представляющего собой цепь одинаковых глюкозных звеньев, но не катализирует гидролиз сахарозы, молекула которой составлена из глюкозного и фруктозного фрагментов.
Согласно общепринятым представлениям о механизме ферментативного катализа, субстрат S и фермент F находятся в равновесии с очень быстро образующимся фермент-субстратным комплексом FS, который сравнительно медленно распадается на продукт реакции P с выделением свободного фермента; т.о., стадия распада фермент-субстратного комплекса на продукты реакции является скоростьопределяющей (лимитирующей).
F + S <––> FS ––> F + P
Исследование зависимости скорости ферментативной реакции от концентрации субстрата при неизменной концентрации фермента показали, что с увеличением концентрации субстрата скорость реакции сначала увеличивается, а затем перестает изменяться (рис. 2.12) и зависимость скорости реакции от концентрации субстрата описывается следующим уравнением:
 (II.45)
(II.45)
Здесь Кm – константа Михаэлиса, численно равная концентрации субстрата при V = ½Vmax. Константа Михаэлиса служит мерой сродства между субстратом и ферментом: чем меньше Кm, тем больше их способность к образованию фермент-субстратного комплекса.
Характерной особенностью действия ферментов является также высокая чувствительность активности ферментов к внешним условиям – рН среды и температуре. Ферменты активны лишь в достаточно узком интервале рН и температуры, причем для ферментов характерно наличие в этом интервале максимума активности при некотором оптимальном значении рН или температуры; по обе стороны от этого значения активность ферментов быстро снижается.

Рис. 2.12Зависимость скорости ферментативной реакции от концентрации субстрата.
3 ТЕРМОДИНАМИКА РАСТВОРОВ
Существование абсолютно чистых веществ невозможно – всякое вещество обязательно содержит примеси, или, иными словами, всякая гомогенная система многокомпонентна. Если имеющиеся в веществе примеси в пределах точности описания системы не оказывают влияния на изучаемые свойства, можно считать систему однокомпонентной; в противном случае гомогенную систему считают раствором.
Раствор – гомогенная система, состоящая из двух или более компонентов, состав которой может непрерывно изменяться в некоторых пределах без скачкообразного изменения её свойств.
Раствор может иметь любое агрегатное состояние; соответственно их разделяют на твердые, жидкие и газообразные (последние обычно называют газовыми смесями). Обычно компоненты раствора разделяют на растворитель и растворенное вещество. Как правило, растворителем считают компонент, присутствующий в растворе в преобладающем количестве либо компонент, кристаллизующийся первым при охлаждении раствора; если одним из компонентов раствора является жидкое в чистом виде вещество, а остальными – твердые вещества либо газы, то растворителем считают жидкость. С термодинамической точки зрения это деление компонентов раствора не имеет смысла и носит поэтому условный характер.
Одной из важнейших характеристик раствора является его состав, описываемый с помощью понятия концентрация раствора. Ниже дается определение наиболее распространенных способов выражения концентрации и формулы для пересчета одних концентраций в другие, где индексы А и В относятся соответственно к растворителю и растворенному веществу.
Молярная концентрация С – число молей νВ растворенного вещества в одном литре раствора.
Нормальная концентрация N – число молей эквивалентов растворенного вещества (равное числу молей νВ, умноженному на фактор эквивалентности f) в одном литре раствора.
Моляльная концентрация m – число молей растворенного вещества в одном килограмме растворителя.
Процентная концентрация ω – число граммов растворенного вещества в 100 граммах раствора.
 (III.1)
(III.1)
 (III.2)
(III.2)
 (III.3)
(III.3)
Еще одним способом выражения концентрации является мольная доля X – отношение числа молей данного компонента к общему числу молей всех компонентов в системе.
 (III.4)
(III.4)
3.1 ОБРАЗОВАНИЕ РАСТВОРОВ. РАСТВОРИМОСТЬ
Концентрация компонента в растворе может изменяться от нуля до некоторого максимального значения, называемого растворимостью компонента. Растворимость S – концентрация компонента в насыщенном растворе. Насыщенный раствор – раствор, находящийся в равновесии с растворенным веществом. Величина растворимости характеризует равновесие между двумя фазами, поэтому на неё влияют все факторы, смещающие это равновесие (в соответствии с принципом Ле Шателье – Брауна).
Образование раствора является сложным физико-химическим процессом. Процесс растворения всегда сопровождается увеличением энтропии системы; при образовании растворов часто имеет место выделение либо поглощение теплоты. Теория растворов должна объяснять все эти явления. Исторически сложились два подхода к образованию растворов – физическая теория, основы которой были заложены в XIX веке, и химическая, одним из основоположников которой был Д.И. Менделеев. Физическая теория растворов рассматривает процесс растворения как распределение частиц растворенного вещества между частицами растворителя, предполагая отсутствие какого-либо взаимодействия между ними. Единственной движущей силой такого процесса является увеличение энтропии системы ΔS; какие-либо тепловые или объемные эффекты при растворении отсутствуют (ΔН = 0, ΔV = 0; такие растворы принято называть идеальными). Химическая теория рассматривает процесс растворения как образование смеси неустойчивых химических соединений переменного состава, сопровождающееся тепловым эффектом и изменением объема системы (контракцией), что часто приводит к резкому изменению свойств растворенного вещества (так, растворение бесцветного сульфата меди СuSО4 в воде приводит к образованию окрашенного раствора, из которого выделяется не СuSО4, а голубой кристаллогидрат СuSО4·5Н2О). Современная термодинамика растворов основана на синтезе этих двух подходов.
В общем случае при растворении происходит изменение свойств и растворителя, и растворенного вещества, что обусловлено взаимодействием частиц между собой по различным типам взаимодействия: Ван-дер-Ваальсового (во всех случаях), ион-дипольного (в растворах электролитов в полярных растворителях), специфических взаимодействий (образование водородных или донорно-акцепторных связей). Учет всех этих взаимодействий представляет собой очень сложную задачу. Очевидно, что чем больше концентрация раствора, тем интенсивнее взаимодействие частиц, тем сложнее структура раствора. Поэтому количественная теория разработана только для идеальных растворов, к которым можно отнести газовые растворы и растворы неполярных жидкостей, в которых энергия взаимодействия разнородных частиц EA-B близка к энергиям взаимодействия одинаковых частиц EA-A и EB-B. Идеальными можно считать также бесконечно разбавленные растворы, в которых можно пренебречь взаимодействием частиц растворителя и растворенного вещества между собой. Свойства таких растворов зависят только от концентрации растворенного вещества, но не зависят от его природы.
3.1.1 Растворимость газов в газах
Газообразное состояние вещества характеризуется слабым взаимодействием между частицами и большими расстояниями между ними. Поэтому газы смешиваются в любых соотношениях (при очень высоких давлениях, когда плотность газов приближается к плотности жидкостей, может наблюдаться ограниченная растворимость). Газовые смеси описываются законом Дальтона:
Общее давление газовой смеси равно сумме парциальных давлений всех входящих в неё газов.
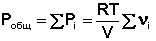 (III.5)
(III.5)
 (III.6)
(III.6)
3.1.2 Растворимость газов в жидкостях
Растворимость газов в жидкостях зависит от ряда факторов: природы газа и жидкости, давления, температуры, концентрации растворенных в жидкости веществ (особенно сильно влияет на растворимость газов концентрация электролитов).
Наибольшее влияние на растворимость газов в жидкостях оказывает природа веществ. Так, в 1 литре воды при t = 18 °С и P = 1 атм. растворяется 0.017 л. азота, 748.8 л. аммиака или 427.8 л. хлороводорода. Аномально высокая растворимость газов в жидкостях обычно обусловливается их специфическим взаимодействием с растворителем – образованием химического соединения (для аммиака) или диссоциацией в растворе на ионы (для хлороводорода). Газы, молекулы которых неполярны, растворяются, как правило, лучше в неполярных жидкостях – и наоборот. Зависимость растворимости газов от давления выражается законом Генри – Дальтона:
Растворимость газа в жидкости прямо пропорциональна его давлению над жидкостью.
 (III.7)
(III.7)
Здесь С – концентрация раствора газа в жидкости, k – коэффициент пропорциональности, зависящий от природы газа. Закон Генри – Дальтона справедлив только для разбавленных растворов при малых давлениях, когда газы можно считать идеальными. Газы, способные к специфическому взаимодействию с растворителем, данному закону не подчиняются.
Растворимость газов в жидкостях существенно зависит от температуры; количественно данная зависимость определяется уравнением Клапейрона – Клаузиуса (здесь X – мольная доля газа в растворе, λ – тепловой эффект растворения 1 моля газа в его насыщенном растворе):
 (III.8)
(III.8)
Как правило, при растворении газа в жидкости выделяется теплота (λ < 0), поэтому с повышением температуры растворимость уменьшается. Растворимость газов в жидкости сильно зависит от концентрации других растворенных веществ. Зависимость растворимости газов от концентрации электролитов в жидкости выражается формулой Сеченова (X и Xo – растворимость газа в чистом растворителе и растворе электролита с концентрацией C):
 (III.9)
(III.9)
3.1.3 Взаимная растворимость жидкостей
В зависимости от природы жидкости могут смешиваться в любых соотношениях (в этом случае говорят о неограниченной взаимной растворимости), быть практически нерастворимыми друг в друге либо обладать ограниченной растворимостью. Рассмотрим последний случай на примере системы анилин – вода. Если смешать примерно равные количества воды и анилина, система будет состоять из двух слоев жидкости; верхний слой – раствор анилина в воде, нижний – раствор воды в анилине. Для каждой температуры оба раствора имеют строго определенный равновесный состав, не зависящий от количества каждого из компонентов.
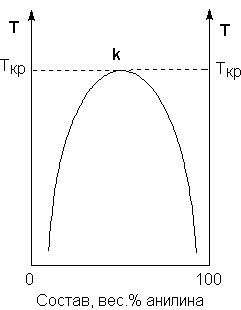
Рис. 3.1 Диаграмма растворимости системы анилин – вода
Зависимость концентрации растворов от температуры принято изображать графически с помощью диаграммы взаимной растворимости. Эта диаграмма для системы анилин-вода приведена на рис. 3.1. Область под кривой – это область расслаивания жидкостей. Повышение температуры приводит к увеличению концентрации каждого из растворов (увеличению взаимной растворимости), и при некоторой температуре, называемой критической температурой расслоения (Ткр на рис. 3.1) взаимная растворимость воды и анилина становится неограниченной. Система анилин – вода относится к т.н. системам с верхней критической температурой расслоения; существуют также и системы, для которых повышение температуры приводит к уменьшению взаимной растворимости компонентов.
3.1.4 Растворимость твердых веществ в жидкостях
Растворимость твердых веществ в жидкостях определяется природой веществ и, как правило, существенно зависит от температуры; сведения о растворимости твердых тел целиком основаны на опытных данных. Качественным обобщением экспериментальных данных по растворимости является принцип "подобное в подобном": полярные растворители хорошо растворяют полярные вещества и плохо – неполярные, и наоборот.

Рис. 3.2 Кривые растворимости некоторых солей в воде.
1 – КNО3, 2 – Nа2SО4·10Н2О, 3 – Nа2SО4, 4 – Ва(NО3)2
Зависимость растворимости S от температуры обычно изображают графически в виде кривых растворимости (рис. 3.2). Поскольку теплота растворения твердых веществ в жидкостях может быть как положительной, так и отрицательной, растворимость при увеличении температуры может увеличиваться либо уменьшаться (согласно принципу Ле Шателье – Брауна).
3.2 РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ
3.2.1 Давление насыщенного пара разбавленных растворов
Представим, что в равновесную систему жидкость А – пар введено некоторое вещество В. При образовании раствора мольная доля растворителя XА становится меньше единицы; равновесие в соответствии с принципом Ле Шателье – Брауна смещается в сторону конденсации вещества А, т.е. в сторону уменьшения давления насыщенного пара РА. Очевидно, что, чем меньше мольная доля компонента А в растворе, тем меньше парциальное давление его насыщенных паров над раствором. Для некоторых растворов выполняется следующая закономерность, называемая первым законом Рауля:
Парциальное давление насыщенного пара компонента раствора прямо пропорционально его мольной доле в растворе, причем коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом.
 (III.10)
(III.10)
Поскольку сумма мольных долей всех компонентов раствора равна единице, для бинарного раствора, состоящего из компонентов А и В легко получить следующее соотношение, также являющееся формулировкой первого закона Рауля:
 (III.11)
(III.11)
Относительное понижение давления пара растворителя над раствором равно мольной доле растворенного вещества и не зависит от природы растворенного вещества.
Растворы, для которых выполняется закон Рауля, называют идеальными растворами. Идеальными при любых концентрациях являются растворы, компоненты которых близки по физическим и химическим свойствам (оптические изомеры, гомологи и т.п.) и образование которых не сопровождается объёмными и тепловыми эффектами. В этом случае силы межмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором. Растворы, компоненты которых существенно различаются по физическим и химическим свойствам, подчиняются закону Рауля лишь в области бесконечно малых концентраций.
3.2.2 Давление пара идеальных и реальных растворов
Если компоненты бинарного (состоящего из двух компонентов) раствора летучи, то пар над раствором будет содержать оба компонента (относительное содержание компонентов в парах будет, как правило, отличаться от содержания их в растворе – пар относительно богаче компонентом, температура кипения которого ниже). Рассмотрим бинарный раствор, состоящий из компонентов А и В, неограниченно растворимых друг в друге. Общее давление пара, согласно первому закону Рауля, равно
 (III.12)
(III.12)
Таким образом, для идеальных бинарных растворов зависимость общего и парциального давления насыщенного пара от состава раствора, выраженного в мольных долях компонента В, является линейной при любых концентрациях (рис.3.3). К таким системам относятся, например, системы бензол – толуол, гексан – гептан, смеси изомерных углеводородов и др.

Рис. 3.3 Зависимость парциальных и общего давлений пара идеального раствора от концентрации
Для реальных растворов данные зависимости являются криволинейными. Если молекулы данного компонента взаимодействуют друг с другом сильнее, чем с молекулами другого компонента, то истинные парциальные давления паров над смесью будут больше, чем вычисленные по первому закону Рауля (положительные отклонения). Если же однородные частицы взаимодействуют друг с другом слабее, чем разнородные, парциальные давления паров компонентов будут меньше вычисленных (отрицательные отклонения). Реальные растворы с положительными отклонениями давления пара образуются из чистых компонентов с поглощением теплоты (ΔНраств > 0), растворы с отрицательными отклонениями образуются с выделением теплоты (ΔНраств < 0).

Рис. 3.4 Зависимость парциальных и общего давлений пара идеальных (штриховая линия) и реальных (сплошная линия) бинарных растворов от состава при положительных (слева) и отрицательных (справа) отклонениях от закона Рауля.
3.2.3 Температура кристаллизации разбавленных растворов
Раствор в отличие от чистой жидкости не отвердевает целиком при постоянной температуре; при некоторой температуре, называемой температурой начала кристаллизации, начинают выделяться кристаллы растворителя и по мере кристаллизации температура раствора понижается (поэтому под температурой замерзания раствора всегда понимают именно температуру начала кристаллизации). Замерзание растворов можно охарактеризовать величиной понижения температуры замерзания ΔТзам, равной разности между температурой замерзания чистого растворителя T°зам и температурой начала кристаллизации раствора Tзам:
 (III.13)
(III.13)
Рассмотрим Р – T диаграмму состояния растворителя и растворов различной концентрации (рис. 3.5), на которой кривая ОF есть зависимость давления пара над твердым растворителем, а кривые ОА, ВС, DE – зависимости давления пара над чистым растворителем и растворами с возрастающими концентрациями соответственно. Кристаллы растворителя будут находиться в равновесии с раствором только тогда, когда давление насыщенного пара над кристаллами и над раствором одинаково. Поскольку давление пара растворителя над раствором всегда ниже, чем над чистым растворителем, температура, отвечающая этому условию, всегда будет более низкой, чем температура замерзания чистого растворителя. При этом понижение температуры замерзания раствора ΔTзам не зависит от природы растворенного вещества и определяется лишь соотношением числа частиц растворителя и растворенного вещества.

Рис. 3.5 Понижение температуры замерзания разбавленных растворов
Можно показать, что понижение температуры замерзания раствора ΔTзам прямо пропорционально моляльной концентрации раствора:
 (III.14)
(III.14)
Уравнение (III.14) называют вторым законом Рауля. Коэффициент пропорциональности K – криоскопическая постоянная растворителя – определяется природой растворителя.
3.2.4 Температура кипения разбавленных растворов
Температура кипения растворов нелетучего вещества всегда выше, чем температура кипения чистого растворителя при том же давлении. Рассмотрим Р – T диаграмму состояния растворителя и растворов различной концентрации (рис.3.5). Любая жидкость – растворитель или раствор – кипит при той температуре, при которой давление насыщенного пара становится равным внешнему давлению. Соответственно температуры, при которых изобара Р = 1 атм. пересечет кривые ОА, ВС и DE, представляющие собой зависимости давления пара над чистым растворителем и растворами с возрастающими концентрациями соответственно, будут температурами кипения этих жидкостей (рис. 3.6).
Повышение температуры кипения растворов нелетучих веществ ΔTк = Tк – T°к пропорционально понижению давления насыщенного пара и, следовательно, прямо пропорционально моляльной концентрации раствора. Коэффициент пропорциональности E есть эбулиоскопическая постоянная растворителя, не зависящая от природы растворенного вещества.
 (III.15)
(III.15)

Рис. 3.6 Повышение температуры кипения разбавленных растворов
Т.о., второй закон Рауля можно в наиболее общем виде сформулировать следующим образом:
Понижение температуры замерзания и повышение температуры кипения разбавленного раствора нелетучего вещества прямо пропорционально моляльной концентрации раствора и не зависит от природы растворенного вещества.
Второй закон Рауля является следствием из первого; данный закон справедлив только для бесконечно разбавленных растворов. Коэффициенты пропорциональности в уравнениях (III.14 – III.15) – эбулиоскопическая и криоскопическая константы – имеют физический смысл соответственно повышения температуры кипения и понижения температуры замерзания растворов с моляльной концентрацией, равной 1 моль/кг. Однако, поскольку такие растворы не являются бесконечно разбавленными, эбулиоскопическая и криоскопическая константы не могут быть непосредственно определены и относятся поэтому к числу т.н. экстраполяционных констант.
3.2.5 Осмотическое давление разбавленных растворов
Если разделить два раствора с различной концентрацией полупроницаемой перегородкой, пропускающей молекулы растворителя, но препятствующей переходу частиц растворённого вещества, будет наблюдаться явление самопроизвольного перехода растворителя через мембрану из менее концентрированного раствора в более концентрированный – осмос. Осмотические свойства раствора количественно характеризуются величиной осмотического давления. Давление, которое необходимо приложить к раствору, чтобы предотвратить перемещение растворителя в раствор через мембрану, разделяющую раствор и чистый растворитель, есть осмотическое давление π. Осмотическое давление идеальных растворов линейно зависит от температуры и молярной концентрации раствора С и может быть рассчитано по уравнению (III.16):
 (III.16)
(III.16)
Уравнение (III.16) есть т.н. принцип Вант-Гоффа:
осмотическое давление идеального раствора равно тому давлению, которое оказывало бы растворенное вещество, если бы оно, находясь в газообразном состоянии при той же температуре, занимало бы тот же объем, который занимает раствор.
Осмос играет важнейшую роль в процессах жизнедеятельности животных и растений, поскольку клеточная плазматическая мембрана является полупроницаемой. Осмос обусловливает поднятие воды по стеблю растений, рост клетки и многие другие явления.
Рассмотрим роль осмоса в водном режиме растительной клетки. Осмотическое давление жидкости, контактирующей с клеткой, может быть больше, меньше либо равно осмотическому давлению внутриклеточной жидкости. Соответственно выделяют гипертонические, гипотонические и изотонические растворы.
Если клетка находится в контакте с гипертоническим раствором, вода выходит из неё путём осмоса через плазматическую мембрану. Протопласт (живое содержимое клетки) при этом уменьшается в объёме, сморщивается и в конце концов отстаёт от клеточной стенки. Этот процесс называют плазмолизом. Процесс плазмолиза обычно обратим.
Если клетку поместить в чистую воду или гипотонический раствор, вода путём осмоса поступает в клетку; протопласт при этом увеличивается в объёме и оказывает давление на сравнительно жёсткую клеточную стенку. Этот процесс называется тургором. Тургорное давление препятствует дальнейшему поступлению воды в клетку. Именно тургорное давление поддерживает стебли растений в вертикальном положении, придаёт растениям прочность и устойчивость.
Изотонические растворы не оказывают влияния на водный режим клетки.
У животных клеток нет клеточной стенки, поэтому они более чувствительны к осмотическому давлению жидкости, в которой находятся. Животные клетки имеют систему защиты, основанную на осморегуляции; организм животного стремится поддерживать осмотическое давление всех тканевых жидкостей на постоянном уровне. Например, осмотическое давление крови человека – 800 000 Н/м2. Такое же осмотическое давление имеет 0,9 %-ный раствор хлорида натрия. Физиологический раствор, изотоничный крови, широко применяется в медицине.
3.2.6 Понятие активности растворенного вещества
Если концентрация растворенного вещества не превышает 0.1 моль/л, раствор неэлектролита обычно считают разбавленным. В таких растворах взаимодействие между молекулами растворителя существенно преобладает над взаимодействием между молекулами растворителя и растворенного вещества, поэтому последним обычно можно пренебречь. В случае более концентрированных растворов такое приближение неправомерно и для формального учета взаимодействия частиц растворителя и растворенного вещества, а также частиц растворенного вещества между собой, вводится эмпирическая величина, заменяющая концентрацию – активность (эффективная концентрация) а, связанная с концентрацией через коэффициент активности f, который является мерой отклонения свойств реального раствора от идеального:
 (III.17)
(III.17)
Как правило, коэффициент активности меньше единицы (при малых концентрациях считают f = 1 и а = С). Необходимо отметить, что активность компонента не прямо пропорциональна его концентрации – коэффициент активности уменьшается с увеличением концентрации.
3.3 РАСТВОРЫ ЭЛЕКТРОЛИТОВ
3.3.1 Теория электролитической диссоциации
Законы Рауля и принцип Вант-Гоффа не выполняются для растворов (даже бесконечно разбавленных), которые проводят электрический ток – растворов электролитов. Обобщая экспериментальные данные, Я.Г. Вант-Гофф пришел к выводу, что растворы электролитов всегда ведут себя так, будто они содержат больше частиц растворенного вещества, чем следует из аналитической концентрации: повышение температуры кипения, понижение температуры замерзания, осмотическое давление для них всегда больше, чем вычисленные. Для учета этих отклонений Вант-Гофф внес в уравнение (III.16) для растворов электролитов поправку – изотонический коэффициент i:
 (III.18)
(III.18)
Аналогичная поправка вносится в законы Рауля, и изотонический коэффициент определяется следующим образом:
 (III.19)
(III.19)
Изотонический коэффициент для растворов электролитов всегда больше единицы, причем с разбавлением раствора i возрастает до некоторого целочисленного значения.
Для объяснения особенностей свойств растворов электролитов С. Аррениус предложил теорию электролитической диссоциации, основывающуюся на следующих постулатах:
1. Электролиты в растворах распадаются на ионы – диссоциируют;
2. Диссоциация является обратимым равновесным процессом;
3. Силы взаимодействия ионов с молекулами растворителя и друг с другом малы (т.е. растворы являются идеальными).
Диссоциация электролитов в растворе происходит под действием полярных молекул растворителя; наличие ионов в растворе предопределяет его электропроводность. Для оценки полноты диссоциации в теории электролитической диссоциации вводится понятие степень диссоциации α, которая равна отношению числа молекул n, распавшихся на ионы, к общему числу молекул N:
 (III.20)
(III.20)
Величина степени диссоциации зависит от природы растворителя и растворенного вещества, концентрации раствора и температуры. По величине степени диссоциации электролиты подразделяются на три группы: сильные (α ≥ 0.7), средней силы (0.3 < α < 0.7) и слабые (α ≤ 0.3). К сильным электролитам относятся почти все соли (кроме Рb(СН3СОО)2, НgСl2, СdСl2), большинство неорганических кислот и щелочей; к слабым – все органические кислоты, вода, NН4ОН, Н2S и т.д. Электролитами средней силы являются некоторые неорганические кислоты: НF, НСN, Н3PO4.
3.3.2 Слабые электролиты. Константа диссоциации
Процесс диссоциации слабых электролитов является обратимым и в системе существует динамическое равновесие, которое может быть описано константой равновесия, выраженной через концентрации образующихся ионов и непродиссоциировавших молекул, называемой константой диссоциации. Для некоторого электролита, распадающегося в растворе на ионы в соответствии с уравнением:
АaВb <––> aАx- + bВy+
константа диссоциации выразится следующим соотношением:
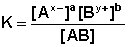 (III.21)
(III.21)
Для бинарного (распадающегося на два иона) электролита выражение (III.21) можно переписать в виде (III.21a):
 (III.21a)
(III.21a)
Поскольку концентрация каждого иона для бинарного электролита равна произведению степени диссоциации α на общую концентрацию электролита С, выражение (III.21a) в этом случае можно переписать следующим образом:
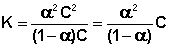 (III.22)
(III.22)
Для разбавленных растворов можно считать, что (1 – α) = 1. Тогда получаем:
 (III.23)
(III.23)  (III.24)
(III.24)
Т.о., степень диссоциации слабого электролита обратно пропорциональна концентрации и прямо пропорциональна разбавлению раствора; выражение (III.24) называют законом разбавления Оствальда. Степень диссоциации слабого электролита можно связать с изотоническим коэффициентом. Будем считать, что из N молекул электролита продиссоциировало n молекул, образовав νn ионов (ν – число ионов, на которое диссоциирует молекула). Поскольку изотонический коэффициент показывает, во сколько раз общее число молекул и ионов в растворе больше числа молекул до диссоциации, получаем:
 (III.25)
(III.25)
 (III.26)
(III.26)
Соотношение (III.26) дает возможность, экспериментально определив изотонический коэффициент раствора, рассчитать степень диссоциации слабого электролита.
3.3.3 Сильные электролиты
Предположение Аррениуса о том, что в растворе сильного электролита также существует динамическое равновесие между молекулами и ионами, как и у слабых электролитов, оказалось ошибочным. Экспериментальные исследования показали, что, во-первых, величина константы диссоциации сильного электролита зависит от концентрации (т.е. к растворам сильных электролитов неприменим закон действующих масс) и, во-вторых, никакими методами не удалось обнаружить в растворах сильных электролитов непродиссоциировавшие молекулы. Это позволило сделать вывод, что сильные электролиты в растворах любых концентраций полностью диссоциируют на ионы и, следовательно, закономерности, полученные для слабых электролитов, не могут применяться к сильным электролитам без соответствующих поправок.
Качественная теория сильных электролитов была разработана П. Дебаем и Г. Хюккелем (1923). Для сильных электролитов, полностью диссоциирующих на ионы, даже при малых концентрациях растворов энергия электростатического взаимодействия между ионами достаточно велика, и пренебречь этим взаимодействием нельзя. Взаимодействие противоположно и одноименно заряженных ионов (соответственно притяжение и отталкивание) приводит к тому, что вблизи каждого иона находятся преимущественно ионы с противоположным зарядом, образующие т.н. ионную атмосферу. Радиус ионной атмосферы сравнительно велик, поэтому ионные атмосферы соседних ионов пересекаются; кроме того, каждый ион окружен дипольными молекулами растворителя – сольватной оболочкой. Т.о., в растворе сильного электролита возникает подобие пространственной структуры, что ограничивает свободу перемещения ионов и приводит к изменению свойств раствора в том же направлении, как действовало бы уменьшение степени диссоциации. Поэтому, определяя степень диссоциации раствора сильного электролита, получают т.н. кажущуюся степень диссоциации, т.е. величину α с поправкой на межионное взаимодействие. Чем выше концентрация раствора, тем сильнее взаимодействие ионов, тем меньше и кажущаяся степень диссоциации сильного электролита.
Количественные расчеты характеристик растворов сильных электролитов осуществляют с помощью понятий активности электролита аэ и активностей катионов и анионов а+ и а- соответственно, которые равны произведению коэффициента активности на концентрацию:
 ;
;  ;
;  (III.27)
(III.27)
Для бинарного электролита средняя активность электролита связана с активностями ионов соотношением (III.28); подобным же образом связан средний коэффициент активности с ионными:
 (III.28)
(III.28)
 (III.29)
(III.29)
Дебаем и Хюккелем был разработан метод расчета среднего коэффициента активности сильного электролита. Для бинарного электролита уравнение имеет следующий вид:
 (III.30)
(III.30)
Здесь z – заряд иона, для которого рассчитывается коэффициент активности, I – т.н. ионная сила раствора: некоторый параметр, который одновременно учитывает молярную концентрацию и заряд всех имеющихся в растворе ионов. Ионная сила раствора равна полусумме концентраций всех ионов, умноженных на квадрат их заряда:
 (III.31)
(III.31)
Теория Дебая – Хюккеля применима только при концентрациях, не превышающих 0.05 моль/л. Для более концентрированных растворов сильных электролитов количественной теории не существует.
3.4 ЭЛЕКТРОПРОВОДНОСТЬ РАСТВОРОВ ЭЛЕКТРОЛИТОВ
3.4.1 Удельная электропроводность растворов электролитов
Электрический ток есть упорядоченное перемещение заряженных частиц. Растворы электролитов обладают ионной проводимостью (являются т.н. проводниками второго рода), т.е. электропроводность растворов электролитов обусловлена перемещением ионов в электрическом поле (в отличие от электронной проводимости проводников первого рода).
Величина преимущественного передвижения иона в направлении одного из электродов при прохождении тока через раствор отнесённая к градиенту потенциала 1 В/см, есть абсолютная скорость движения иона. Абсолютные скорости движения ионов имеют величины порядка 0,0005 – 0,003 см2/(В·с). Абсолютные скорости движения катионов U+ и анионов U– различаются; это приводит к тому, что ионы разных знаков переносят разные количества электричества.
Всякий проводник, по которому течет ток, представляет для него определенное сопротивление R, которое, согласно закону Ома, прямо пропорционально длине проводника l и обратно пропорционально площади сечения S; коэффициентом пропорциональности является удельное сопротивление материала ρ – сопротивление проводника, имеющего длину 1 см и сечение 1 см2:
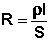 , Ом (III.32)
, Ом (III.32)
В качестве количественной меры способности раствора электролита проводить электрический ток используют обычно удельную электропроводность κ (каппа) – величину, обратную удельному сопротивлению (т.е. величину, обратную сопротивлению столба раствора между электродами площадью 1 см2, находящимися на расстоянии 1 см):
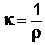 , Ом-1см-1 (III.33)
, Ом-1см-1 (III.33)
Величина удельной электропроводности электролита зависит от ряда факторов: природы электролита, температуры, концентрации раствора. Удельная электропроводность растворов электролитов (в отличие от электропроводности проводников первого рода) с увеличением температуры возрастает, что вызвано увеличением скорости движения ионов за счет понижения вязкости раствора и уменьшения сольватированности ионов. Зависимость удельной электропроводности от концентрации раствора представлена на рис. 3.7.

Рис. 3.7 Зависимость удельной электропроводности электролитов от концентрации
(1 – H2SO4, 2 – KOH, 3 – CH3COOH)
Как видно из рисунка, с увеличением концентрации удельная электропроводность растворов сначала возрастает, достигая некоторого максимального значения, затем начинает уменьшаться. Эта зависимость очень чётко выражена для сильных электролитов и значительно хуже для слабых. Наличие максимума на кривых объясняется тем, что в разбавленных растворах сильных электролитов скорость движения ионов мало зависит от концентрации, и κ сначала растет почти прямо пропорционально числу ионов; с ростом концентрации усиливается взаимодействие ионов, что уменьшает скорость их движения. Для слабых электролитов наличие максимума на кривой обусловлено тем, что с ростом концентрации уменьшается степень диссоциации, и при достижении определенной концентрации число ионов в растворе начинает увеличиваться медленнее, чем концентрация. Для учета влияния на электрическую проводимость растворов электролитов их концентрации и взаимодействия между ионами введено понятие молярной электропроводности раствора.
3.4.2 Молярная электропроводность растворов электролитов
Молярная электропроводность раствора λ есть величина, обратная сопротивлению раствора, содержащего 1 моль растворенного вещества и помещенного между электродами, расположенными на расстоянии 1 см друг от друга. С удельной электропроводностью κ и молярной концентрацией раствора С молярная электропроводность связана следующим соотношением:
 , Ом-1см2моль-1 (III.34)
, Ом-1см2моль-1 (III.34)
Молярная электропроводность как сильных, так и слабых электролитов увеличивается с уменьшением конц
Дата добавления: 2014-12-03; просмотров: 315; Мы поможем в написании вашей работы!; Нарушение авторских прав |