КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Тема 4. Криминалистическая фотография, звуко- , видеозапись
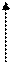 |
Ангиотензин I


Антиренин
Ангиотензин II Ангиотензин III
 |  |
Ангиотензиназы
Рис. 37.1. Компоненты ренин-ангиотензиновой системы.
В настоящее время к почечной ренин-ангиотензиной системе добавились и другие истинные ренин-продуцирующие системы, обнаруженные и в других органах. Такие рениноподобные вещества, называемые изоренинами, были обнаружены в подчелюстной железе, матке и плаценте, амниотической жидкости, лимфе, а также в гипофизе и эпифизе.
Секреция ренина на уровне юкстагломерулярного аппарата зависит от многих факторов:
- почечного кровотока;
- количества натрия в крови и моче;
- общего объёма крови;
- состояния вегетативной нервной системы.
Роль ренин-ангиотензиновой системы. Установлено, что активный ангиотензин обладает двумя доказанными действиями: сосудосуживающим и альдостеронвысвобождающим. Помимо этих эффектов, ангиотензин выполняет ряд медиаторных функций, посредниками которых являются катехоламины и альдостерон. Ренин-изоренин-ангиотензиновая система действует как единая гормональная система. Различные раздражители (ишемия, гипонатриемия, гиповолемия, пониженное артериальное давление и др.) стимулируют высвобождение тканевого ренина и изоренина с увеличением содержания ангиотензина, что приводит к сосудистым и метаболическим эффектам. Благодаря сосудосуживающему, альдостеронвысвобождающему и стимулирующему действиям, ренин-ангиотензиновая система приводит к увеличению периферического сопротивления сосудов, к гиперволемии, вызывая, таким образом, артериальную гипертензию. Увеличение артериального давления может быть транзиторным, компенсаторным механизмом в случае понижения артериального давления, при гиповолемии, при ишемии почек, либо как персистирующее явление, поддерживаемое морфофункциональным повреждением почечной паренхимы.
Помимо последствий сосудосуживающих эффектов, при различных гипертензивных синдромах ренин-ангиотензивная система проявляется и нарушением нормальной секреции альдостерона. Активность плазматического ренина возрастает при различных формах вторичного гиперальдостеронизма, сопровождающимися повышенным артериальным давлением и отёками.
Эритропоэтин. Вследствие гипоксии, в системном кровотоке появляется фактор со стимулирующим действием на эритропоэз. Этот фактор был назван стимулирующим фактором эритропоэза (СФЭ) или эритропоэтин. Впоследствии установили, что в больших количествах эритропоэтин вырабатывается на уровне почки.
Относительно механизма образования и эффектов эритропоэтина была выявлена аналогия с ренин-ангиотензиновой системой и плазмакининами. Эритропоэтин вырабатывается, также, как ангиотензин и биологически активные плазмакинины, вседствие ферментативного действия на плазматический глобулин.
Также доказано, что эритропоэтин происходит из группы плазматических, биологически активных полипептидов. Стимулирование эритропоэза эритропоэтином приводит к увеличению объёма крови посредством роста количества эритроцитов, в то время как ангиотензин и плазмакинины вовлечены в регуляцию объёма плазмы.

 Гипоксия
Гипоксия
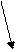
 Почка Печень
Почка Печень
Эритрогенин Плазматический глобулин
 |  |
Эритропоэтин
 |
Гематопоэтические органы
Рис. 37.2. Образование и эффекты эритропоэтина.
Почечный сосудорасширяющий фактор
В мозговом веществе почки располагаются интерстициальные клетки, наполненные липидами. Эти клетки имеют все свойства, характерные для секреторнх клеток, и находятся во взаимосвязи с vasa rectae и канальцами. То, что соответствующие клетки имеют секреторные свойства, а их наполнение липидами варьирует, привлекло внимание относительно возможности выработки и высвобождения этими клетками «антигипертензивного реномедулярного липида».
Простагландины
На уровне почки были обнаружены многие типы простагландинов. Химическая структура этих веществ имеет в своей основе ненасыщенную жирную кислоту с 20 атомами углерода – простаноевую. Было продемонстрировано, что медуллин состоит из компонентов липидного происхождения, среди которых есть PGE2 и PGF1 – альфа и PGA2. Среди них самым выраженным вазомоторным действием обладает PGE2. Впоследствии установили, что в мозговом веществе почки синтезируются и другие типы простагландинов, например, PGF2 – альфа.
Синтетизируемые на уровне почки простагландины ферментативно катаболизируются 15-гидроксипростагландиндегидрогеназой.
Простагландины действуют в очень маленьких дозах, как правило, в нанограммах. Их действие различно для разных органов. Почка подвергается влиянию простагландинов как на уровне сосудистого, так и канальциевого полюса. На уровне почечной артерии простагландины могут индуцировать вазодиллятацию, уменьшая сопротивление почечной сосудистой сети, что приводит к увеличению перфузии, к увеличению образования мочи и экскреции ионов натрия. Такими же эффектами обладают PGA1 , PGA2. Чаще всего, гемодинамические эффекты простагландинов на уровне почки селективны, они вызывают перераспределение почечного кровотока к коре и уменьшению интенсивности кровотока мозгового слоя.
На уровне канальцев простагландины определяют уменьшение реабсорбции натрия и повышение его выведения с мочой.
Кининообразующие ферменты
Кининогены (например, калликреиноген), выделенные в кровь, действуют на плазматический глобулин (кининоген) с образованием биологически активных веществ типа брадикинина. Сразу после образования, плазмакинины быстро инактивируются карбоксипептидазами (кининазами) путём отщепления аргинина из полипептидной цепи.
 Калликреиноген
Калликреиноген
Калликреин
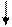
 Кининоген
Кининоген
 Плазмакинины Киназы
Плазмакинины Киназы
Рис. 37.3. Схема калликреин-кининовой системы
Образуясь в крови, плазмакинины проявляют множественные эффекты, включая расширение сосудов и увеличение проницаемости капилляров. На уровне почек эти эффекты представлены в полном объеме. Описаны явления истинной конкуренции между вазоактивными полипептидами на уровне капилляров почек и общего кровообращения, в результате чего устанавливается динамическое равновесие между ренин-ангиотензиновой и плазмакининовой системами.
В зависимости от преобладания протеолитической активности калликреина или ренина на плазматический альфа-2-глобулин, результатом будет синтез плазмакининов (брадикинина) или ангиотензина с антагонистическим действием на сосуды, что обеспечивает сохранение клубочкового кровотока в гомеостатических пределах.
37.6.Почечная недостаточность.
Почечная недостаточность может развиться в результате патологических процессов в самих почках, равно как и при некоторых преренальных и субренальных процессах.
Почечная недостаточность представляет собой временое или персистирующее нарушение функций почек и сопровождается общими метаболическими, водно-электролитными, кислотно-щелочными и циркуляторными дисгомеостазами. В зависимости от клинического течения почечная недостаточность может быть острой и хронической.
Острая почечная недостаточность ( ОПН )
Этиология и патогенез ОПН. Острая почечная недостаточность может быть следствием как почечных патологических процессов (с повреждением паренхимы почки), так и внепочечных патологических процессов (преренальных и постренальных).
Среди преренальных этиологических факторов выделяются те, которые нарушают почечную перфузию и, как следствие, клубочковую фильтрацию без первичных структурных изменений почек. В основе нарушений почечных функций стоит развивающаяся ишемия почек. При выраженной и длительной гипоперфузии могут развиться и повреждения почечной паренхимы различных степеней - при этом на последствия действия преренальных этиологических факторов накладываются и интраренальные.
Первая группа преренальных этиологических факторов ОПН включает:
- выраженную гиповолемию при массивных кровотечениях, дегидратации (диарея, неукратимая рвота, пищеварительные фистулы, злоупотребление диуретиками, несахарный и сахарный диабет, потоотделение, ожоги, водянка);
- уменьшение сердечного выброса при остром инфаркте миокарда, эмболии легочной артерии, миокардитах и др.;
- тяжёлая системная вазодиллятация при шоке различной этиологии, септицемии, при лечении гипотензивными средствами;
- длительная почечная вазоконстрикция при лечении адреналином и эрготамином.
Уменьшение почечного кровотока, которое сопровождает все перечисленные патологические процессы, ведёт к перераспределению внутрипочечного кровотока в мозговое вещество с ишемией коркового слоя, что ведёт к уменьшению клубочковой фильтрации. Восстановление почечного кровотока может нормализовать фильтрацию, а длительная почечная гипоперфузия вызывает повреждения клубочков и острый некроз канальцев. В этой стадии восстановление почечного кровотока не приводит к нормализации функций почек. Чем длительнее ишемия, тем необратимее ишемические повреждения канальцев. Ишемические повреждения почек развиваются из-за энергетического дефицита, избыточного тока ионов Са в клетку, внутриклеточного ацидоза, нарушения активности ионных насосов, дисфункций митохондрий. Некротизированные клетки канальцев могут «забивать» канальцы и, таким образом, основной ишемический механизм повреждения может осложниться вторичным обструктивным механизмом.
К внутрипочечным этиологическим факторам развития острой почечной недостаточности относят факторы с нефротоксическим действием – экзогенные и эндогенные токсины. Нефротоксический эффект прямо пропорционален концентрации и длительности действия этиологического фактора.
К экзогенным нефротоксическим веществам относят органические растворители, соли тяжёлых металлов, токсины грибов, рентгеноконтрастные вещества, химиотерапевтические вещества, антибиотики, анальгетики.
Нефротоксические вещества вызывают повреждения эпителия канальцев, более выраженные на уровне собирательных трубочек, что вызывает массивную ретродиффузию клубочкового фильтрата с появлением олигурии. Десквамированные клетки и клеточный детрит можгут закупорить канальцы и, таким образом, способствовать присоединению вторичного обструктивного механизма повреждения. Обструктивный механизм объясняет олигоанурию как проявление почечной недостаточности, вызванную механическим препятствием току мочи. Уменьшение клубочковой фильтрации является также результатом увеличением гидростатического давления в мочевыводящих канальцах и капсуле Bowman, которое сравнивается с гломерулярным гидростатическим давлением и препятствует процессу фильтрации.
Среди эндогенных веществ, которые могут привести к ОПН, отмечают: свободный гемоглобин при усиленном гемолизе; миоглобин из поврежденных скелетных мышц, который фильтруется через клубочек и затем осаждается в почечных канальцах, закупоривая их; осаждение в канальцах кристаллов (кальций, оксалаты, мочевая кислота) с таким же эффектом.
К постренальным факторам развития острой почечной недостаточности, которые блокируют мочевые пути и препятствуют нормальному выведению мочи, относят: опухоли, камни, стенозы, сгустки крови. Основным механизмом возникновения ОПН в таком случае является обструкция мочевых путей с последующим увеличением внутрикапсулярного давления и уменьшением клубочковой фильтрации. Обструкция мочевых путей приводит к рефлекторному спазму капилляров коркового слоя, сопровождающийся корковой ишемией и снижением фильтрации.
Проявления острой почечной недостаточности.
Главными синдромами ОПН любой этиологии являются: мочевой, гуморальный и клинический.
Главными проявлениями, составляющими мочевой синдром, являются нарушение диуреза и способности почек к разведению и концентрации мочи.
Нарушения диуреза в периоде разгара ОПН проявляются олигурией (менее 350-400 мл мочи за 24 часа, достигая при тяжёлых формах 50 мл/24 часа - анурия). Олигурия может длиться до 2-3 недель, но иногда и более месяца, период, во время которого может развиться канальцевый некроз, гломерулонефрит, васкулит. После фазы олигоанурии диурез постепенно увеличивается, наступает фаза полиурии, развивающаяся как результат снижения чувствительности регенерированных клеток канальцев к АДГ. Тяжёлая полиурия ведёт к серьёзным водно-электролитным нарушениям, иногда с летальными последствиями.
Нарушения функций разведения и концентрации мочи проявляются изостенурией, уменьшенной концентрацией электролитов в конечной моче из-за нарушения противоточно-множительного механизма, который способствует процессу концентрирования мочи. Постепенно, способность тубулярного аппарата к разведению мочи восстанавливается, таким образом, полиурия становится гипостенурической, а затем восстанавливается и концентрационная функция.
Гуморальный синдром включает водно-электролитные и кислотно-щелочные нарушения. Нарушения способности почек поддерживать химические константы внутренней сруды в нормальных пределах ведёт к ряду водно-электролитных и кислотно- щелочных дисгомеостазов на фоне задержки азотистых метаболитов.
Задержка азотистых метаболитов происходит как из-за уменьшения клубочковой фильтрации, так и из-за белкового гиперкатаболизма. Отмечается увеличение концентрации мочевины в крови при неосложнённых формах - на 10-20 мг% каждые сутки, а при гиперкатаболических формах – на 20-100мг%. Концентрация креатинина, мочевой кислоты и аминокислот растет медленнее.
Водно-электролитные нарушения вызваны олигоанурией и проявляются глобальной гипергидратацией (если потребление воды превышает её потери) или внеклеточной гипергидратацией (если потребление натрия превышет его потери). Реже развивается дегидратация в фазах постанурической полиурии.
Нарушения электролитного баланса:
- концентрация натрия часто снижена (гипонатриемия и гипоосмолярность) -
следствие чрезмерного потребления воды;
- изменения концентрации ионов хлора аналогичны изменениям натрия, за исключением потерь через пищеварительный тракт;
- концентрация ионов калия увеличена (гиперкалиемия), даже при отсутствии экзогенных источников калия; при формах, осложненных септицемией, гемолизом, при синдроме длительного сдавливания тканей уровень калиемии больше и может определить нарушения ритма и проводимости на уровне миокарда с характерными изменениями на ЭКГ;
- уровень кальциемии, как правило, снижен из-за гиперфосфатемии, гипоальбуминемии и нарушений почечного гидроксилирования витамина D, с уменьшением кишечной абсорбции кальция;
- концентрация магния умеренно повышена;
- концентрация фосфатов и сульфатов повышены.
Кислотно-щелочные нарушения характеризуются метаболическим ацидозом из-за нарушения почечных механизмов обеспечивающих поддержание концентрации ионов водорода на постоянном уровне. Секреция ионов Н в почечных канальцах значительно снижается, параллельно уменьшается и количество бикарбонатов в плазме крови, которые используются для нейтрализации кислых метаболитов.
Клинический синдром включает различные нарушения жизненных функций
Дыхательные дисфункции представлены, преимущественно, нарушениями дыхательного ритма (дыхание Cheyne -Stokes ), частоты и амплитуды дыхания (дыхание Kussmaul,; развивающимися при гиперазотемии и метаболическом ацидозе.
Сердечно-сосудистые симптомы проявляются артериальной гипертонией в случае водно-солевой задержки, нарушениями сердечного ритма вследствие дизэлектролитемии или сопутствующего миокардита, иногда сердечной недостаточности.
Симптомы нарушений функций пищеварительной системы встречаются у 50% пациентов и представлены тошнотой, рвотой, диарреей, анорексией, а при тяжёлых формах - меленой, как следствие стрессогенных язв.
Нейро-психические симптомы являются результатом действия азотистых токсических продуктов, метаболического ацидоза или отёка мозга и проявляются астенией, головными болями, тонико-клоническими судорогами.
Гематологические нарушения представлены анемией вследствие угнетения эритропоэза, а также усилениия гемолиза, лейкоцитозом (даже при отсутствии инфекций) и тромбоцитопенией.
Постоянно присутствующие нарушения гемостаза развиваются из-за качественных дефектов тромбоцитов, недостатка тромбоцитогенеза и нарушений синтеза некоторых факторов свертывания крови, приводящими к кровотечениям. Эти нарушения усугубляют течение ОПН.
Хроническая почечная недостаточность (ХПН)
Хроническая почечная недостаточность является результатом медленно прогрессирующего уменьшения массы функционирующих нефронов.
Этиология ХПН. Большинство врожденных или приобретенных хронических нефропатий могут привести к ХПН. Самыми частыми причинами ХПН являются:
- первичные и вторичные повреждения клубочков - воспаление, некроз, склерозирование, аутоиммунные повреждения, коллагенозы (системная красная волчанка и др.), опухолевые и метаболические поражения (диабетический гломерулосклероз);
- тубуло-интерстициальные поражения, представленные хроническим пиелонефритом, метаболическими нефропатиями (гиперурикемия или гиперкальциемия), нефропатиями при хронических отравлениях лекарствами или солями тяжёлых металлов, при хронической обструктивной нефропатии;
- почечные сосудистые поражения - тромбоз почечных вен, стеноз почечных артерий, нефроангиосклероз, узелковый периартериит;
- обширные деструктивные процессы в почечной паренхиме (опухоли почек, врожденные аномалии – поликистозы почек), специфические хронические воспаления.
Патогенез ХПН. Патогенез почечных дисфункций при ХПН объясняется различными патогенетическими теориями: теорией «анархии нефронов» и теорией «интактного нефрона».
Согласно теории «анархии нефронов», функции почек обеспечиваются всеми нефронами, независимо от степени повреждения, т.е., наблюдается функциональная гетерогенность нефронов.
Согласно теории «интактного нефрона», принятой многими специалистами, основные функции почек поддерживаются нефронами оставшимися морфофункционально интактными. Благодаря функционирующим нефронам определённый период времени клубочково-канальцевое равновесие поддерживается в нормальных пределах – компенсаторная фаза. По мере уменьшения количества функционирующих нефронов, нагрузка, выполняемая каждым нефроном в отдельности, растёт. Когда оставшиеся нефроны не в состоянии обеспечивать функции почек, наступает фаза декомпенсации с прогрессивным течением до стадии уремии.
Клиническое развитие ХПН отражает стадии компенсации и декомпенсации.
Первая стадия - стадия полной компенсации, часто является клинически латентной, функциональные клубочково-канальциевые пробы незначительно изменены, но гомеостаз не нарушен. В этом периоде оставшиеся функциональные нефроны составляют более 50% всей популяции нефронов.
Вторая стадия характеризуется компенсированной гиперазотемией. Поддержание гомеостаза осуществляется посредством «компенсаторной полиурии». Уменьшение количества функционирующих нефронов менее 25% приводит к водно- электролитным и кислотно-щелочным нарушениям.
Третья стадия - стадия декомпесации, предполагает уменьшение популяции функционирующих нефронов менее 25% и проявляется выраженной гиперазотемией, морфофункциональными изменениями с полиморфной клинической картиной.
Четвертая стадия - терминальная уремия - предполагает уменьшение популяции функционирующих нефронов менее 10% и характеризуется олигоанурией, значительной гиперазотемией, выраженными нарушениями гомеостаза с полиморфной клинической картиной. В этой стадии выживание возможно лишь при постоянном проведении гемодиализа или трансплантации почки.
Синдромы ХПН имеют различный патогенез.
Мочевой синдром. Во второй стадии развития ХПН появляется полиурия как результат осмотического диуреза, который обусловлен повышенной концентрацией в первичной моче электролитов и мочевины. Способность канальцев к разведению и концентрации мочи нарушена и выделяемая моча становится изостенурической, с относительной плотностью равной плотности депротеинизированной плазмы. Постепенно, по мере уменьшения количества функционирующих нефронов, полиурия переходит в олигурию, а в конечной стадии - в анурию.
Метаболико-гуморальный синдром включает компенсированную гиперазотемию с постепенным повышением концентрации креатинина, мочевой кислоты, которая может обусловить появление интерстициальной нефропатии, что приводит к ухудшению почечных функций. Уремические токсины, образовавшиеся при нормальном или изменённом метаболизме, накапливаются в высоких концентрациях и оказывают токсические эффекты. Уремические токсины представлены гуанидинсукциновой кислотой, полиамидами (путресцин), фенолами, феноловыми производными, пептидами со средней молекулярной массой и даже повышенной концентрацией паратгормона.
Нарушение водно-электролитного баланса. При ХПН может развиться дегидратация (в фазе компенсаторной полиурии) или гипергидратация (в олиго-анурической фазе). Концентрация электролитов в плазме крови меняется в зависимости от стадии ХПН – натриемия поддерживается в нормальных пределах до третьей стадии ХПН, в конечных стадиях происходит задержка натрия в крови, или может установиться гипонатриемия из-за внепочечных потерь натрия.
Калиемия может повыситься в конечных стадиях ХПН, особенно в при выраженном катаболизме (лихорадка, инфекции) или при гемолизе и других клеточных повреждениях. Нарушения фосфорно-кальциевого равновесия ведут к развитию почечной остеодистрофии. Основными патогенетическими механизмами остеодистрофии являются нарушение метаболизма витамина D3, гиперфосфатемия, метаболический ацидоз и задержка токсических метаболитов.
Нарушения кислотно–щелочного равновесия проявляется экскреторным ацидозом вследствие снижения экскреции ионов водорода на уровне почечных канальцев или вследствие потери бикарбонатов.
Нарушения метаболизма и трансмембранного транспорта. На поздних стадиях ХПН наблюдается снижение активности Na-K-АТФ-азы как следствие накопления уремических токсинов в организме. Нарушается трансмембранный транспорт электролитов, что приводит к избыточному внутриклеточному накоплению натрия, прекращению почечной канальцевой реабсорбции натрия и повышению нервно-мышечной возбудимости.
Промежуточный метаболизм также нарушается. Установлено увеличение инсулинемии (из-за уменьшенной почечной экскреции) и возможное развитие гипогликемии. В этих случаях наблюдается увеличение плазматической концентрации СТГ и глюкагона, нарушения трансмембраного транспорта глюкозы с тенденцией к гипергликемии.
Нарушение жирового обмена. Из-за снижения активности некоторых ферментов (липопротеинлипазы, печеночной липазы), происходит снижение катаболизма липопротеинов, одновременно с последующим увеличением их уровня в сыворотке крови и развитием вторичной гиперлипопротеинемии (особенно III и IV типа ). Наблюдается развитие раннего выраженного атеросклероза, который не зависит от возраста и пола.
Изменения белкового обмена состоят в усилении катаболизма с последующей гипераминоацидемией.
Клинический синдром.
Изменения в дыхательной системе при развитии ХПН представлены уремическим отёком лёгких, обусловленным увеличением проницаемости лёгочных капилляров и недостаточностью левого желудочка. Будучи хроничексим процессом, отек лёгких может привести к отложению фибрина в интерстиции легких с развитием фиброза. При ХПН часто поражается плевра (серозные и геморрагические плевриты).
Сердечно-сосудистые изменения, наряду с инфекционными осложнениями, представляют собой одну из главных причин смертности при ХПН. Ишемическая кардиомиопатия часто развивается из-за усиленного атерогенеза.
Уремическая кардиомиопатия, возникающая, в особенности, из-за уремических токсинов, наряду с ишемической кардиопатией, способствует развитию аритмий - атривентрикулярных блокад различной степени, вплоть до развития сердечной недостаточности.
Перикардиальные отложения мочевины или мочевой кислоты обусловливают воспаление этой серозной оболочки (перикардит).
Артериальная гипертония (АГ) развивается у 70% больных с тяжёлой формой ХПН. Основными патогенетическими факторами развития АГ являются гипернатриемия, гиперволемия и повышение периферического сопротивления сосудов.
Нарушения пищеварительной системы охватывают все сегменты пишеварительного тракта и, в большей мере, вызваны мочевиной, которая выделяется через кишечник и под действием местной микробной флоры превращается в токсические вещества (карбонат и карбомат азота).
Появляются язвы слизистой оболочки ротовой полости и языка с нарушением вкусовой чувствительности и появлением запаха аммиака в выдыхаемом воздухе.
Костно-суставные изменения являются выражением нарушений фосфорно-кальциевого обмена и проявляются в форме почечной остеодистрофии, характеризующейся нарушениями роста и развития костей, остеомаляцией, остеопорозом, остеосклерозом или фиброзным остеитом. Гиперурикемия может привести к повреждению суставов подобно подагре.
Гематологические изменения проявляются гипорегенераторной анемией, степень которой зависит от заболевания, вызвавшего ХПН. Анемия возникает из-за недостатка синтеза эритропоэтина, токсического угнетения гематопоэтической системы, мальнутриции, дефицита железа и фолатов, а также вследствие гемолиза или кровотечений.
Изменения гемостаза проявляется кровоточивостью (экхимозы, пурпура, эпистаксис, кровотечения из пищеварительного тракта и др.), вызванными тромбоцитарными нарушениями и недостатчным синтезом факторов коагуляции.
Иммунологические изменения характеризуются нарушением функции специфической и неспецифической защиты, что приводит к увеличению частоты инфекционных осложнений.
Нервно-психические нарушения появляются вследствии уремической интоксикации, кислотно-щелочных нарушений, отёка мозга и представлены астенией, апатией, нарушениями речи и зрения, раздражимостью, признаками периферической нейропатии и др.
Уремическая кома представляет собой терминальную клиническую стадию с различными проявлениями.
Считается, что ответственным за клинические проявления уремии является токсическое действие ароматических веществ, задерживающихся в организме, водно-электролитные и кислотно-щелочные нарушения (дегидратация, гипергидратация, ацидоз).
Библиография
1. Abram E. S. The pain clinic manual. J. B. Lippincott Co. 1990, 180 p.
2. Arseni C., Chimion D. Lichidul cefalo-rahidian. Editura didactică şi pedagogica. Bucureşti, 1979, 271 p.
3. Arseni C., Popoviciu L. Metode de neurofiziologie clinică. Ed. Medicală, Bucureşti, 178 p.
4. Badescu M., Ciocoiu M. Compendiu de fiziopatologie specială. Editura Vasiliana, Iaşi, 2001, 371p.
5. Barbu R., Nedelcu I. Rinichiul. Fiziopatologie clinică. Bucureşti, 1988, 315 p.
6. Berceanu S. Hematologie clinică. Ed.Med. Bucureşti, 1977, 943 p.
7. Botnaru V. Hipertensiunea arterială: aspecte clinice. Chişinău, 1996, 192 p.
8. Botnaru V. Bolile cardiovacsulare: aspecte de diagnostic. Chişinău, 1997, 349 p.
9. Botnaru V. Bolile aparatului respirator. Chişinău, 2001, 637 p.
10. Cazacu P. Fiziopatologie - 1000 teste la computer. C.E.P. Medicina al USMF, Chişinău, 1998, 315 p.
11. Cobâleanschi L., Cazacu P., Lutan V., Ţuşco V. Dicţionar explicativ fiziopatologic român-rus-francez. Chişinău, Ştiinţa, 1993, 270 p.
12. Corcimaru I.T. Hematologie clinică. Chisinau, 2001, 298 p.
13. Corcimaru I.T. Anemiile. Chişinău, 2003, p.159.
14. Coliţa D. Anemiile. Clasificare. Tratat de medicină internă. Partea I. Editura medicală, Bucureşti, 1997, 556 p.
15. Cristea I. Terapia durerii. Editura Medicală, Bucureşti, 1996, 246 p.
16. Cucuianu M, Trif,I, Cucuianu A. Hemostaza. Editura Dacia, Cluj-Napoca,1994,.380 p.
17. Dinu M. Colev V, Bădescu M. Fiziopatologie (curs). Tipografia U.MF., Iaşi, 1988. 210 p.
18. Dorofteiu Mircea. Fiziologie: coordonarea organismului uman. Editura Argonaut, Cluj-Napoca, 1992, 279 p.
19. Gherasim L. Medicina internă. Vol. III Bolile digestive, hepatice şi pancreatice. Bucureşti, Editura medicală, 2002. 1209 p.
20. Gherasim L. Medicina internă. Vol. II. Bolile cardiovasculare, metabolice. Bucureşti, 1996, 1356 p.
21. Grigoriu G., Puşcariu T. Hematopoieza. Tratat de medicină internă.
Partea I. Editura medicală, Bucureşti, 1997, 943 p.
22. Grosu A. Aritmiile cardiace. Chişinău, 1999, 263 p.
23. Guyton A. Fiziologie. Ediţia a 5-a. Bucureşti, 1997, 587 p.
24. Hăulică I. Fiziologie umană. Editura medicală, Bucureşti, 1989,1214p.
25. Hossu T., Marcu J., Munteanu N. Anemiile. Tratat de medicină internă. Partea I. Editura medicală, Bucureşti, 1997, 943 p.
26. Iarovoi A., Cazacu P., Cobâleanschi L., Pitel E. Introducere în imunopatologie. Chişinău, 1995, 90 p.
27. Păun R. Tratat de medicină internă.Bolili cardiovscculare. Partea II. Bucureşti, 1989, 840 p.
28. Răileanu –Moţoiu I. Limfopoieza. Tratat de medicină internă. Partea I. Editura medicală, Bucureşti, 1997, 241 p.
29. Rusnac T. Maladiile nefro- urinare la copil. Tipografia centrală. Chişinau.2001, 280 p.
30. Saragea M. Fiziopatologie vol. II. Editura Academiei Republicii Socialiste România, 1982, 1188 p.
31. Teodorescu Exarcu I. Fiziologia şi fiziopatologia sistemului nervos. Editura Medicală, Bucureşti, 1978, 1071 p.
32. Teodorescu Exarcu I. Fiziologia şi fiziopatologia digestiei.Editura Medicală, Bucureşti, 1982, 706 p.
33. Vâlcu A. Eritropoeza. Tratat de medicină internă. Partea I. Editura medicală, Bucureşti, 1997, 943 p.
34. Aдо А.Д. Патологическая физиология. Москва, "Триада-Х", 2001, 457c.
35. Баркаган З.С. Геморагические заболевания и синдромы. Москва, «Медицина» 1988, 500 c.
36. Бурчинский Г. И. Клиническая гастро-энтероглогия. Киев, Здоровья, 1979, 635 с.
37. Вандер A. Физиология почек (перевод с англ.). Санкт Петербург. 2000. 190 с.
38. Вейн А. М., Хехт К. Сон человека. Физиология и патология. Москва, Медицина, 1989, 266 c.
39. Воробьев А.И. Руководство по гематологии том.1. Москва Медицина» 1985, 442 c.
40. Воробьев А.И. Руководство по гематологии том.2. Москва «Медицина» 1985, 366 c.
41. Галперин Э.И. и соавт. Недостаточность печени. М., «Медицина»1978, 327 c.
42. Гаврилов О.К., Козинец Г.И. Черняк Н.Б. Клетки костного мозга и периферической крови. Москва «Медицина» 1985, 277c.
43. Гаврилов О.К. Проблемы и гипотезы в учении о свертывании крови. Москва «Медицина» 1981, 328 с.
44. Глебов Р.Н., Крыжановский Г.Н. Функциональная биохимия синапсов. Москва, 1978, 324 c.
45. Годухин О.В. Модуляция синаптической передачи в мозге. Москва, 1987, 157 c.
46. Денхема М. Дж., Чанарина И.( M.J.Denhan, I. Chanarin). Болезни крови у пожилых. Москва «Медицина» 1987, 350 c.
47. Зайчик А.Ш., Чурилов Л. П. Основы общей патологии. "Элби - СПБ". Специальная литература, Санкт-Петербург, 1999, 618с.
48. Зайчик А.Ш., Чурилов Л. П. Основы патохимии. "Элби - СПБ". Специальная литература, Санкт-Петербург, 2001, 687с.
49. Зайчик А.Ш., Чурилов Л. П. : Механизмы развития болезней и синдромовю "Элби - СПБ". Специальная литература, Санкт-Петербург, 2002, 618с.
50. Зайко Н.Н. Патологическая физиология. Киев, "Вища школа", 1996, 647c.
51. Ковалева Л.Г. Острые лейкозы. Москва «Медицина» 1990, 269 c.
52. Калиничева В.И. Анемии у детей. Ленинград, «Медицина» 1983. 358 c.
53. Леонович А. Л. Актуальные вопросы невропатологии. Минск, 1990, 206 c.
54. Литвицкий П.Ф. Патофизиология. Курс лекций. Москва, 1995,750c.
55. Литвицкий П.Ф. Патофизиология, том 2. Москва, ГЭОТАР-МЕД, 2002, 808 c.
56. Логинов А.С. Блок Ю.Е. Хронические гепатиты и цирозы печени. Москва Медицина»1987, 272 c.
57. Петелин Л.С. Ретикулярная формация ствола мозга и синдромы ее поражения. Москва,1982. 265 c.
58. Павлов А.Д., Морщакова Е.Ф. Регуляция эритропоэза. Москва «Медицина» 1987, 233 c.
59. Полак Дж.М. Физиология и патофизиология желудочно-кишечного тракта. (перевод с английского). Москва, “Медицина” 1989, 495c.
60. Подымова С.Д. Болезни печени. М., «Медицина», 1984, 480 с.
61. Файнштейн Ф.Э., Козинец Г.И и др., Болезни системы крови. Ташкент, «Медицина» 1987, 650 c.
62. Фербенкс В.Ф.(Virgil F. Fairbanks ). Железо. Метаболизм и клинические нарушения. В кн. Современная гематология и онкология. Москва «Медицина» 1987, 448 c.
63. Филлип Дж. Файалкоу ( Philip J. Fialkov). Миелопролиферативные заболевания. В кн: Современная гематология и онкология. Москва, «Медицина» 1985, 305 c.
64. Ханбабян М.В. Норадренергические механизмы мозга. Санкт –Петербург, 1981, 122c.
65. Шанин В.Ю. Клиническая патофизиология. Санкт-Петербург. Специальная Литература, 1998, 659c.
66. Шиффман Ф. Дж.Патофизиология крови. М., СПб. Бином – Невский диалект, 2000, 448 c.
67. Шмидт Р. Ф. Физиология человека. Том 3 Москва, 1986.
68. Шмидт Р. Основы сенсорной физиологии. Москва, 1984, 286 c.
Тема 4. Криминалистическая фотография, звуко- , видеозапись
Занятие второе – практическое (2 часа)
План
1. Устройство фотоаппарата, его основные узлы, детали, механизмы. Цифровой фотоаппарат.
2. Основные характеристики объектива.
3. Понятие и назначение диафрагмы, шкалы глубины резкости,
шкалы расстояний.
Дата добавления: 2014-12-23; просмотров: 360; Мы поможем в написании вашей работы!; Нарушение авторских прав |