КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Спектроскопические методы анализа
Все спектроскопические методы основаны на взаимодействии атомов, молекул или ионов, входящих в состав анализируемого вещества, с электромагнитным излучением. Это взаимодействие проявляется в поглощении или испускании фотонов (квантов). В зависимости от характера взаимодействия пробы с электромагнитным излучением выделяют две группы методов - эмиссионные и абсорбционные.В зависимости от того, какие частицы формируют аналитиче- ский сигнал, различают методы атомной спектроскопии и методы молекулярной спектроскопии.

|
В эмиссионных методах анализируемая проба в результате ее возбуждения излучает фотоны (кванты). Важнейшие эмиссионные методы - атомно-эмиссионный спектральный анализ(АЭС) и люминесцентный анализ.
В абсорбционных методахизлучениепостороннего источника пропускают через пробу, при этом часть квантов избирательно поглощается атомами или молекулами. Важнейшие методы этой группы - атомно-абсорбционный анализ (ААС) и молекулярно-абсорбционная спектроскопия растворов. Последний метод обычно называют спектрофотометрией или фотометрическим анализом. Абсорбционные методы, как и эмиссионные, используют и для обнаружения, и для количественного определения веществ.
Кроме спектроскопических, известны и другие методы анализа, основанные на оптических явлениях. В частности, в нефелометрии используют эффект рассеяния света, врефрактометрии - преломление светового потока; в поляриметрии – вращение плоскости поляризации. Эти оптические методы к числу спектроскопических не относят, поскольку они не связаны с поглощением или излучением квантов.
34. Спектры атомов. Основные и возбужденные состояния атомов, характеристики состояний. Энергетические переходы. Правила отбора. Законы испускания и поглощения
Атомные спектры возникают при переходах между уровнями энергии внешних электронов атома и наблюдаются в видимой, ультрафиолетовой и близкой инфракрасной областях. Такими спектрами обладают как нейтральные, так и ионизованные атомы; их часто называют соответственно дуговыми и искровыми спектрами (нейтральные атомы легко возбуждаются и дают спектры испускания в электрических дугах, а положительные ионы возбуждаются труднее и дают спектры испускания преимущественно в искровых электрических разрядах). Спектры ионизованных атомов смещены по отношению к спектрам нейтральных атомов в область больших частот, т. е. в ультрафиолетовую область. Это смещение тем больше, чем выше кратность ионизации атома — чем больше электронов он потерял. Спектры нейтрального атома и его последовательных ионов обозначают в спектроскопии цифрами I, II, III, ... В реально наблюдаемых спектрах часто присутствуют одновременно линии нейтрального и ионизованных атомов; так говорят, например, о линиях FeI, FeII, FeIII в спектре железа, соответствующих Fe, Fe+, Fe2+.
Линии А. с. образуют закономерные группы, называются спектральными сериями. Промежутки между линиями в серии убывают в сторону коротких длин волн, и линии сходятся к границе серии. Наиболее прост спектр атома водорода. Волновые числа линий его спектра с огромной точностью определяются формулой Бальмера:
1/l = R(1/n21 - 1/n22),
где n1 и n2 значения главного квантового числа для уровней энергии, между которыми происходит квантовый переход (см. Атом,рис. 1, б). Значение n1 = 1, 2, 3, ... определяет серию, а значение n2 = n1 + 1, n1 + 2, n1 + 3,... определяет отдельные линии данной серии;R — Ридберга постоянная (выраженная в волновых числах). При n1 = 1 получается серия Лаймана, лежащая в далёкой ультрафиолетовой области спектра, при n1 = 2 — серия Бальмера, линии которой расположены в видимой и близкой ультрафиолетовой областях. Серии Пашена (n1 = 3), Брэкета (n1 = 4), Пфаунда (n1 = 5), Хамфри (n1 = 6) лежат в инфракрасной области спектра. Аналогичными спектрами, только с увеличенным в Z2 раз масштабом (Z — атомный номер), обладают водородоподобные ионы Не+, Li2+, ... (cпектры HeII, LiIII, ...).
Для атомов с двумя или несколькими внешними электронами спектры значительно усложняются, что обусловлено взаимодействием электронов. А. с. особенно сложны для атомов с заполняющимися d- и f-оболочками; число линий доходит до многих тысяч, и уже нельзя обнаружить простых серий, аналогичных сериям в спектрах водорода и щелочных металлов. Однако и в сложных спектрах можно установить определённые закономерности в расположении линий, произвести систематику спектра и определить схему уровней энергии.
В А. с. проявляются не все переходы между уровнями энергии данного атома или иона, а лишь вполне определённые, допускаемые (разрешенные) т. н. отбора правилами, зависящими от характеристик уровней энергии. В случае одного внешнего электрона возможны лишь переходы, для которых азимутальное квантовое число l увеличивается или уменьшается на 1; правило отбора имеет вид: Dl = ±1. В результате s-yровни (l = 0) комбинируют с р-уровнями (l = 1), р-уровни — с d-yровнями (l = 2) и т. д., что определяет возможные спектральные серии для атомов щелочных металлов, частный случай которых представляет главная серия Na (переходы 3s ® np, где n =3, 4, 5, ...); другие переходы этим правилом отбора запрещены. Для многоэлектронных атомов правила отбора имеют более сложный вид.
Количественной характеристикой разрешенного оптического перехода является его вероятность (см. Вероятность перехода),определяющая, как часто этот переход может происходить; вероятность запрещенных переходов равна нулю. От вероятностей переходов зависят интенсивности спектральных линий. В простейших случаях вероятности переходов для А. с. могут быть рассчитаны по методамквантовой механики.
Наряду с изучением А. с. для свободных атомов значительный интерес представляет исследование изменений в А. с. при внешних воздействиях на атомы. Под действием внешнего магнитного или электрического поля происходит расщепление уровней энергии атома и соответствующее расщепление спектральных линий
Исследование А. с. сыграло важную роль в развитии представлений о строении атома (см. Атомная физика). Методы, основанные на изучении А. с., очень широко распространены в различных областях науки и техники. А. с. позволяют определить ряд весьма важных характеристик атомов и получить ценные сведения о строении электронных оболочек атома. Чрезвычайно существенно применение А. с. в эмиссионном спектральном анализе (по А. с. испускания), который благодаря высокой чувствительности, быстроте и универсальности завоевал прочное место в металлургии, горнорудной промышленности, машиностроении и во многих других отраслях народного хозяйства; наряду с эмиссионным спектральным анализом успешно применяют и абсорбционный спектральный анализ (по А. с. поглощения).
35. Спектры молекул; их особенности. Основные законы поглощения электромагнитного излучения (Бугера) и закон излучения (Ломакина-Шейбе). Связь аналитического сигнала с концентрацией определяемого соединения.
Закон Бугера - Ламберта - Берасвязывает уменьшение интенсивности света, прошедшего через слой светопоглощающего вещества, с концентрацией вещества и толщиной слоя. Чтобы учесть потери света на отражение и рассеяние, сравнивают интенсивности света, прошедшего через исследуемый раствор и растворитель При одинаковой толщине слоя в кюветах из одинакового материала, содержащих один и тот же растворитель, потери на отражение и рассеяние света будет зависеть от концентрации вещества.
Уменьшение интенсивности света, прошедшего через раствор,характеризуется коэффициентом пропускания (или просто пропусканием) T: T= I / I0, где I и I0 — соответственно интенсивности света, прошедшего через раствор и растворитель.
Взятый с обратным знаком логарифм T называется оптической плотностью A:
-lg T= -lg (I / I0 )=lg (I 0/ I)=A.
Уменьшение интенсивности света при прохождении его через раствор подчиняется закону Бугера-Ламберта-Бера: I=I0 10-e lc , или I / I0=10-e lc ,или -lg T=A=e l c (1)
где e – молярный коэффициент поглощения; l – толщина светопоглощающего слоя;c –концентрация раствора.
Физический смысл e становится ясным, если принять I=1 см и c=1 моль/л, тогда A=e . Следовательно,молярный коэффициент поглощения равен оптической плотности одномолярного раствора при толщине слоя 1 см.
Оптическая плотность раствора, содержащего несколько окрашенных веществ, обладает, свойством аддитивности, которое иногда называют законом аддитивности светопоглощения. В соответствии с этим законом поглощение света каким-либо веществом не зависит от присутствия в растворе других веществ. При наличии в растворе нескольких окрашенных веществ каждое из них будет давать свой аддитивный вклад в
Ограничения и условия применимости закона Бугера-Ламберта–Бера:
1. Закон справедлив для монохроматического света. Чтобы отметить это ограничение, в уравнение (1) вводят индексы и записывают его в виде: Al =e l l c. (2)
Индекс l указывает, что величины A и e относятся к монохроматическому излучению с длиной волны l .
2. Коэффициент e в уравнении (1) зависит от показаеля преломления среды. Если концентрация раствора сравнительно невелика, его показатель преломления остается таким же, каким он был у чистого растворителя, и отклонений от закона по этой причине не наблюдается.
3. Температура при измерениях должна оставаться постоянной хотя бы в пределах нескольких градусов.
4. Пучок света должен бытьпараллельным.
5. Уравнение (1) соблюдается только для систем, в которыхсветопоглощающими центрами являются частицы лишь одного сорта. Если при изменении концентрации будет изменяться природа этих частиц вследствие, например, кислотно-основного взаимодействия, полимеризации, диссоциации и т. д., то зависимость A от с не будет оставаться линейной, так как молярный коэффициент поглощения вновь образующихся и исходных частиц не будет в общем случае одинаковым.
Например, при разбавлении раствора дихромата калия происходит не просто уменьшение концентрации иона дихромата, а протекают процессы химического взаимодействия:
Cr2O2-7 +H2O= 2HCrO-4= 2CrO2-4+2H+
Вместо дихромат-ионов в растворе появляются гидрохромат- и хромат-ионы. Зависимость оптической плотности от общей концентрации хрома в растворе не будет линейной.
36. Аппаратура. Способы монохроматизации лучистой энергии. Классификация спектральных приборов.
Закон Бугера - Ламберта - Берасвязывает уменьшение интенсивности света, прошедшего через слой светопоглощающего вещества, с концентрацией вещества и толщиной слоя. Чтобы учесть потери света на отражение и рассеяние, сравнивают интенсивности света, прошедшего через исследуемый раствор и растворитель При одинаковой толщине слоя в кюветах из одинакового материала, содержащих один и тот же растворитель, потери на отражение и рассеяние света будет зависеть от концентрации вещества.
Уменьшение интенсивности света, прошедшего через раствор,характеризуется коэффициентом пропускания (или просто пропусканием) T: T= I / I0, где I и I0 — соответственно интенсивности света, прошедшего через раствор и растворитель.
Взятый с обратным знаком логарифм T называется оптической плотностью A:
-lg T= -lg (I / I0 )=lg (I 0/ I)=A.
Уменьшение интенсивности света при прохождении его через раствор подчиняется закону Бугера-Ламберта-Бера: I=I0 10-e lc , или I / I0=10-e lc ,или -lg T=A=e l c (1)
где e – молярный коэффициент поглощения; l – толщина светопоглощающего слоя;c –концентрация раствора.
Физический смысл e становится ясным, если принять I=1 см и c=1 моль/л, тогда A=e . Следовательно,молярный коэффициент поглощения равен оптической плотности одномолярного раствора при толщине слоя 1 см.
Оптическая плотность раствора, содержащего несколько окрашенных веществ, обладает, свойством аддитивности, которое иногда называют законом аддитивности светопоглощения. В соответствии с этим законом поглощение света каким-либо веществом не зависит от присутствия в растворе других веществ. При наличии в растворе нескольких окрашенных веществ каждое из них будет давать свой аддитивный вклад в
Ограничения и условия применимости закона Бугера-Ламберта–Бера:
1. Закон справедлив для монохроматического света. Чтобы отметить это ограничение, в уравнение (1) вводят индексы и записывают его в виде: Al =e l l c. (2)
Индекс l указывает, что величины A и e относятся к монохроматическому излучению с длиной волны l .
2. Коэффициент e в уравнении (1) зависит от показаеля преломления среды. Если концентрация раствора сравнительно невелика, его показатель преломления остается таким же, каким он был у чистого растворителя, и отклонений от закона по этой причине не наблюдается.
3. Температура при измерениях должна оставаться постоянной хотя бы в пределах нескольких градусов.
4. Пучок света должен бытьпараллельным.
5. Уравнение (1) соблюдается только для систем, в которыхсветопоглощающими центрами являются частицы лишь одного сорта. Если при изменении концентрации будет изменяться природа этих частиц вследствие, например, кислотно-основного взаимодействия, полимеризации, диссоциации и т. д., то зависимость A от с не будет оставаться линейной, так как молярный коэффициент поглощения вновь образующихся и исходных частиц не будет в общем случае одинаковым.
Например, при разбавлении раствора дихромата калия происходит не просто уменьшение концентрации иона дихромата, а протекают процессы химического взаимодействия:
Cr2O2-7 +H2O= 2HCrO-4= 2CrO2-4+2H+
Вместо дихромат-ионов в растворе появляются гидрохромат- и хромат-ионы. Зависимость оптической плотности от общей концентрации хрома в растворе не будет линейной.
37. Методы атомной оптической спектроскопии. Атомно-эмисионный метод. Источники атомизации и возбуждения
Методы атомной спектроскопии основаны на переходах валентных или внутренних электронов атомов из одного состояния в другое.
Наиболее убедительно наличие уровней энергии атома доказано методами атомной спектроскопии, прежде всего благодаря очень высокой точности определения длин волн.
В зависимости от физической природы процесса взаимодействия излучения с веществом методы атомной спектроскопии электромагнитного излучения ( как оптического, так и рентгеновского диапазона) делят на эмиссионные и абсорбционные
В зависимости от используемого диапазона длин волн электромагнитного излучения и природы соответствующих электронных переходов методы атомной спектроскопии делятся на оптические и рентгеновские.
В последние годы в различных областях физики, химии и техники, наряду с исследованиями методами атомной спектроскопии, приобретают все большее значение исследования методами молекулярной спектроскопии. Первым и зачастую наиболее существенным этапом спектроскопических измерений и исследований является расшифровка спектрограмм - установление принадлежности имеющихся в спектре линий и полос тем или иным состояниям атомов и молекул.
Результаты хорошо согласуются с данными метода флуоресцентной атомной спектроскопии, а Сн равен 1 - 2 мг.
Спектроскопические методы подразделяют также на атомные и молекулярные. Это деление для аналитика принципиально, поскольку в методах атомной спектроскопии мы всегда имеем дело с узкими линейчатыми спектрами, а в методах молекулярной спектроскопии - с широкими слабоструктурированными спектрами. И это в конечном итоге определяет возможность их применения в химическом анализе и требования к измерительной аппаратуре - спектральным приборам.
По изучаемым объектам оптическая спектроскопия подразделяется на атомную н молекулярную. Методами атомной спектроскопии определяются элементы, из которых состоит вещество.
Для определения примесей в чистых веществах пригодны высокоселективные методы, обязательно с низким пределом обнаружения. К числу наиболее высокочувствительных методов относится радиоактивационный анализ. Большее значение приобрели методы атомной спектроскопии, основанные на использовании высокочастотной плазмы, прежде всего индуктивно связанной. Низким пределом обнаружения характеризуется атомно-абсорбционный метод с электротермическими источниками ато-мизации.
Объем сведений, приведенных по определению отдельных элементов, зависит от двух основных факторов. По этому признаку, например, определению свинца, железа, ванадия и некоторых других элементов уделялось больше внимания, а определению серебра, висмута, вольфрама - значительно меньше. Так, из-за отсутствия публикаций по определению в нефтепродуктах углерода, водорода, кислорода и азота методами атомной спектроскопии определение этих элементов рассмотрено лишь в гл.
Атомно-эмиссионная спектроскопия (спектрометрия), АЭС или атомно-эмиссионный спектральный анализ — совокупность методов элементного анализа, основанных на изучении спектров испускания свободных атомов и ионов в газовой фазе (см. группу методов оптической спектроскопии). Обычно эмиссионные спектрырегистрируют в наиболее удобной оптической области длин волн от ~200 до ~1000 нм. (Для регистрации спектров в области <200 нм требуется применение вакуумной спектроскопии, чтобы избавиться от поглощения коротковолнового излучения воздухом. Для регистрации спектров в области >1000 нм требуются специальные инфракрасные или микроволновые детекторы.)
АЭС — способ определения элементного состава вещества по оптическим линейчатым спектрам излучения атомов и ионов анализируемой пробы, возбуждаемым в источниках света. В качестве источников света для атомно-эмиссионного анализа используют пламя горелки или различные виды плазмы, включая плазму электрической искры или дуги, плазму лазерной искры, индуктивно-связанную плазму, тлеющий разряд и др.
АЭС — самый распространённый экспрессный высокочувствительный метод идентификации и количественного определения элементов примесей в газообразных, жидких и твердых веществах, в том числе и в высокочистых. Он широко применяется в различных областях науки и техники для контроля промышленного производства, поисках и переработке полезных ископаемых, в биологических, медицинских и экологических исследованиях и т.д. Важным достоинством АЭС по сравнению с другими оптическими спектральными, а также многими химическими и физико-химическими методами анализа, являются возможности бесконтактного, экспрессного, одновременного количественного определения большого числа элементов в широком интервале концентраций с приемлемой точностью при использовании малой массы пробы.
Процесс атомно-эмиссионного спектрального анализа состоит из следующих основных звеньев:
1. Пробоподготовка (подготовка образца)
2. Испарение анализируемой пробы (если она не газообразная);
3. Диссоциация — 1" атомизация её молекул;
4. Возбуждение излучения атомов и ионов элементов пробы;
5. Разложение возбужденного излучения в спектр;
6. Регистрация спектра;
7. Идентификация спектральных линий — с целью установления элементного состава пробы (качественный анализ);
8. Измерение интенсивности аналитических линий элементов пробы, подлежащих количественному определению;
9. Нахождение количественного содержания элементов с помощью установленных предварительно градуировочных зависимостей.
38. Метод эмиссионной спектрометрии пламени. Подготовка пробы к анализу. Пламенные фотометры и спектрофотометры.
Пламенная фотометрия - оптический метод количественного элементного анализа по атомным спектрам испускания. Для получения спектров анализируемое вещество переводят в атомный пар в пламени. Термическая пламенная фотометрия - разновидность атомного эмиссионного спектрального анализа. В этом методе анализируемый раствор в виде аэрозоля вводят в пламя горючей смеси воздуха или N2O с углеводородами (пропаном, бутаном, ацетиленом). При этом растворитель и соли определяемых металлов испаряются и диссоциируют на своб. атомы. Атомы металлов и образовавшиеся в ряде случаев молекулы их оксидов и гидроксидов возбуждаются и излучают световую энергию. Из всего спектра испускания выделяют характерную для определяемого элемента аналит. линию (с помощью светофильтра или монохроматора) и фотоэлектрически измеряют ее интенсивность, которая служит мерой концентрации данного элемента.
С помощью пламенных фотометров метод пламенной фотометрии применяют для определения щелочных, щелочноземельных, а также некоторых других металлов, напр. Ga, In, Tl, Pb, Mn. Пределы обнаружения щелочных металлов составляют 0,1-0,001 мкг/мл, остальных - 0,1-5 мкг/мл; относит, стандартное отклонение 0,02-0,04. Помехи в методе пламенной фотометрии связаны главным образом с нарушением поступления элемента в пламя вследствие образования труднолетучих соединений (напр., интенсивность излучения Ca снижается в присут. H3PO4 и солей Al) и смещением равновесия ионизации металлов в пламени (напр., излучение К усиливается в присут. Pb и Cs). Помехи устраняют выбором подходящих растворов сравнения, буферных растворов, добавлением спец. реактивов, препятствующих образованию труднолетучих соединений и др.
Пламенные (атомно-абсорбционные) спектрофотометры
Пламенные (атомно-абсорбционные) спектрофотометры имеют обычно один-два канала регистрации. Они измеряют интенсивности линий абсорбции (эмиссии, флуоресценции) атомов элементов в пламени специальных горелок или других "атомизаторов". В простых конструкциях аналитические l выделяются узкополосными фильтрами (пламенные фотометры), в приборах более высокого класса применяются полихроматоры или монохроматоры, которые можно переключать на различные длины волн. Приборы данного типа используют в спектральном анализе для определения большинства элементов периодической системы. Они обеспечивают высокую избирательность и чувствительность до 10-14 г.
39. Атомно-абсорбционный метод. Атомизаторы (пламенные и непламенные). Источники излучения , их характеристики.
Атомно-абсорбционная спектроскопия (ААС) – метод количественного анализа, основанный на свойствах атомов поглощать свет с определенной длиной волны (резонансное поглощение). В зависимости от способа получения поглощающего слоя атомов выделяют 4 основных типов техники атомизации:
· пламенная атомизация – испарение и атомизация происходят в пламени (пропан/воздух, ацетилен/воздух, ацетилен/закись азота). Определяемые концентрации элементов в растворах 0,01 – 100 мг/л;
· электротермическая атомизация (ЭТА) – испарение и атомизация пробы происходит в графитовой трубке (графитовой печи), нагреваемой электрическим током до температур 1500 – 3000 °С (в зависимости от свойств определяемого элемента). Определяемые концентрации элементов в растворах 0,01 – 100 мкг/л;
· гидридная техника – в кварцевой ячейке или графитовой печи, нагреваемой электрическим током, происходит разложение газообразных гидридов, образованных в специальном реакторе: MeHxT --> Me + x/2 H2. Данная техника может использоваться для элементов, образующих термически неустойчивые газообразные гидриды (As, Sb, Se, Sn, Te, Pb). Определяемые концентрации элементов в растворах 0,01 – 100 мкг/л;
· метод «холодного пара» - основан на свойстве ртути существовать при нормальных условиях в газовой фазе в виде свободных атомов. Определяемые концентрации ртути в растворах 0,01 – 100 мкг/л.
Методом ААС могут определяться около 60 элементов (в основном металлы и ряд переходных элементов).
Наивысшую чувствительность в ААС имеют приборы с электротермической атомизацией, в которых, в отличие от приборов с пламенной атомизацией, атомизированная проба остается в замкнутом объеме кюветы, а не уносится газовым потоком, тем самым, большее количество атомов пробы поглощают излучение лампы и чувствительность определения возрастает на 2-3 порядка.
Селективность метода обеспечивается двумя основными факторами:
· свойством атомов поглощать свет только с определенной длиной волны (резонансное поглощение) – исключение влияние на аналитический сигнал других атомов, присутствующих в атомизаторе;
· использование корректора неселективного поглощения – учет вклада в измеряемый сигнал, связанного с поглощением света молекулами и твердыми частицами.
В настоящее время в большинстве коммерческих приборов используют дейтериевый (с источником непрерывного спектра) или Зеемановский корректоры неселективного поглощения. Наилучшую селективность (в ААС необходимо выделить поглощение света атомами определяемого элемента на фоне поглощения посторонними молекулами и частицами) обеспечивает применение эффекта Зеемана.
Эффект Зеемана – расщепление атомной спектральной линии во внешнем магнитном поле на несколько компонентов. В простейшем случае расщепление происходит на три компоненты: одну π, совпадающую с максимумом длины волны исходной линии и 2-х +σ и -σ, симметрично смещенных. Расщепление тем больше, чем выше напряженность магнитного поля. При этом π и σ компоненты имеют различную поляризацию. Благодаря этому можно проводить раздельное измерение поглощения π и σ компонент. При измерении в присутствии π компоненты происходит измерение суммарного поглощения – атомного и неселективного, а в ее отсутствие – только неселективного.
40.Молекулярная абсорбционная спектроскопия (спектрофотометрия). Связь химической структуры соединения с абсорбционным спектром.
В основе молекулярного спектрального анализа лежит качественное (идентификация) и количественное определение индивидуальных веществ или вещества в смесях, основанное на избирательном поглощении молекулами (атомами) вещества электромагнитного излучения. Может быть исследовано как известное молекулярное вещество, так и новые молекулы или иные частицы (ионы, радикалы и др.), а также различные конформеры одних и тех же молекул. Информативность метода определяется строгой индивидуальностью спектра молекул, а сочетание методов анализа по нескольким видам спектров еще более увеличивает надежность определения состава анализируемой пробы. Установлены общие закономерности, связывающие спектр вещества с его строением.
Существует несколько молекулярно-абсорбционных методов: ^колориметрический - основанный на визуальном сравнении окраски анализируемого и стандартного раствора одного и того же вещества (подробнее о нем см. в разделе 5.1.4);
S фотоколориметрический - этот метод основан на фотометрическом измерении (с помощью специальных светочувствительных датчиков) .интенсивности прошедшего через вещество интегрального светового потока (свет всех длин волн);
•S спектрофотометрический - в этом методе измеряется (также с помощью специальных светочувствительных датчиков) интенсивность прошедшего через вещество монохроматического светового потока, причем для каждой длины волны излучения проводится отдельное измерение. В последнем случае методы молекулярного спектрального анализа основаны на сравнении измеренных молекулярных спектров исследуемого образца со стандартными спектрами индивидуальных веществ. В настоящее время созданы приборы, совмещенные с компьютером, которые в процессе съемки спекгра, с помощью специальной программы идентифицируют его и выдают результат анализа в виде формулы вещества с указанием его количества в пробе.
Молекулярный спектр является однозначной характеристикой молекулы, определяется ее свойствами в целом и в первую очередь структурой и свойствами входящих в нее атомов. Он возникает при переходе электронов с основных термов молекулярных орбитатей в возбужденные состояния и может наблюдаться в ультрафиолетовой, видимой и ближней инфракрасной областях (120 - 1000 нм). Энергия переходов составляет 120 - 1000 кДж/моль (30 - 240ккал/моль). При поглощении такой энергии одновременно происходит и изменение в колебательно-вращательных состояниях молекулы, вследствие чего электронные спектры представлены широкими полосами. В разбавленных растворах и парах некоторых соединений полосы электронных переходов расщепляются на более узкие (вращательные и колебательные структуры), характеризующие изменение колебательной и вращательной энергий в возбужденном электронном состоянии.
41. Основной закон светопоглощения. Отклонения от закона.
Основной закон светопоглощения (закон Бугера – Ламберта – Бера)
Если на слой раствора толщиной l падает световой поток с интенсивностью Io, и в результате поглощения света веществом, интен-сивность прошедшего светового потока I уменьшается

Схема прохождения света через слой раствора.
то по закону Бугера–Ламберта–Бера:

Это экспоненциальная форма записи основного закона светопоглощения. В количественном анализе более удобно пользоваться логарифмической формой записи этого же закона:
A = εlC.
Графически закон можно представить в виде (рис.46).
Как следует из графика, тангенс угла наклона графика равен произведению εl. Поскольку в условиях эксперимента l = const, то молярный коэффициент поглощения ε характеризует чувствительность методики анализа.


. Зависимость оптической плотности от концентрации раствора.
В количественном анализе чаще всего используется молярная концентрация, в этом случае коэффициент поглощения ε называют молярным. Если же используется массовая концентрация, то коэффициент поглощения ε называют удельным.
Коэффициент поглощения (погашения, экстинкции) ε характеризует способность вещества поглощать свет. Величина ε зависит от длины волны падающего света и природы вещества, в очень незначительной степени – от температуры. Если переход разрешён, то
ε = 103–105.
Условия применимости закона Бугера – Ламберта – Бера:
§ раствор должен быть разбавленным (С < 0,01 моль/л);
§ свет должен быть монохроматическим и параллельным;
§ не должно протекать побочных химических реакций с участием поглощающих частиц;
§ показатель преломления растворов должен быть постоянным;
§ температура должна быть постоянной.
Если эти условия не соблюдаются, то будут наблюдаться отклонения от закона Бугера – Ламберта – Бера (рис.47).
Причины отклонений от закона Бугера – Ламберта – Бера:
1. Истинные – связаны с изменением показателя преломления растворов при изменении концентрации. При малых концентрациях этим пренебрегают.
2. Кажущиеся – химические и инструментальные. Химические причины связаны с протеканием побочных реакций, в результате чего меняется число и природа поглощающих частиц:
§ протолиз;
§ комплексообразование;
§ окислительно-восстановительные реакции;
§ осаждение;
§ ассоциация и диссоциация;
§ гидролиз;
§ таутомерия и др.
За счёт химических причин могут наблюдаться и положительные, и отрицательные отклонения от закона Бугера – Ламберта – Бера.
Инструментальные причины связаны с недостаточной монохроматизацией света, рассеянием и случайным излучением (возвращаясь в исходное состояние, молекула теряет энергию не в виде теплоты, а в виде излучения). За счёт инструментальных причин наблюдаются отрицательные отклонения от закона Бугера – Ламберта – Бера.
Отклонения от закона Бугера – Ламберта - Бера.
42. Монохроматизация излучения. Понятие об истинном и кажущемся молярном коэффициенте поглощения.
Монохроматический свет — это световые колебания одной частоты. Электромагнитная волна одной определённой и строго постоянной частоты. По своей физической природе электромагнитные волны видимого диапазона не отличаются от волн другово диапазонов (инфракрасного, ультрафиолетового, рентгеновского и т. д.), и по отношению к ним также используют термин «монохроматический» («одноцветный»), хотя никакого ощущения цвета эти волны не дают.
А если сказать, что такое Монохроматический свет пару словами — это свет одной длины.
Монохроматический свет получают несколькими способами :
1) Способ : Призматические системы для выделения потока излучения с заданной степенью монохроматичности

2) Способ : Системы на основе дифракционной решетки.
3) Способ : Газоразрядные лампы и другие источники света, в которых происходит преимущественно один электронный переход (например, натриевая лампа, в излучении которой преобладает наиболее яркая линия D или Ртутная лампа).
При испускании света реальными источниками (лампами, лучами, лазерами) происходит множество переходов между различными энергетическими состояниями; поэтому в таком излучении присутствуют волны многих частот. Приборы, с помощью которых из света выделяют узкие спектральные интервалы близкие к Монохроматическому свету, называют «монохроматорами». Чрезвычайно высокая монохроматичность характерна для излучения некоторых типов лазеров (его спектральный интервал может быть значительно уже, чем у линий атомных спектров).
43. Инструментальные погрешности, оптимальный интервал измеряемых значений оптической плотности.
Измеряемые оптические плотности должны лежать в интервале 0 2 - 0 8, так как в этой области относительная ошибка измерения наименьшая.
Измеряемые оптические плотности относительных почернений должны находиться в области нормальных почернений.
Все измеряемые оптические плотности могут укладываться в оптимальный диапазон измерений, а получаемые оценки дисперсий обладают минимальными свойствами.
При увеличении измеряемой оптической плотности ошибки из-за рассеянного света резко возрастают.
Для перехода от измеряемых оптических плотностей к концентрации элемента в растворе при атомно-абсорб-ционных измерениях в пламени или к абсолютным количествам элементов, вводимых в графитовую кювету, применяются градуировочные графики, которые строят по образцам известного состава, чаще всего приготовленным синтетическим путем.
Кюветы подбирают так, чтобы измеряемые оптические плотности укладывались в интервал значений от 0 1 до 1 0 ( о точности измерений величин D см. стр. Если растворы подчиняются закону Бугера - Ламберта, то выбор кюветы облегчается; например, если кювета с диаметром в 2 см непригодна для измерений интенсивности окраски начальных и конечных растворов эталонного ряда, то можно, измерив оптические плотности с кюветой большего или меньшего диаметра, пересчитать на оптические плотности, соответствующие кювете в 2 см, используя прямую пропорциональность между D и /, и нанести их на один и тот же градуиро - вочный график.
Кюветы подбирают так, чтобы измеряемые оптические плотности укладывались в интервал от 0 1 до 1 0 ( о точности измерений величин А см. стр. Если растворы подчиняются закону Бугера - Ламберта, то выбор кюветы облегчается.
При определении очень малых концентраций измеряемая оптическая плотность может быть ниже 0 1, но при этом для обработки экспериментальных результатов необходимо применять методы математической статистики.
Кюветы подбирают так, чтобы измеряемые оптические плотности укладывались в интервал от 0 1 до 1 0 ( о точности измерений величин А см. стр. Если растворы подчиняются закону Бугера - Ламберта, то выбор кюветы облегчается.
При определении очень малых концентраций измеряемая оптическая плотность может быть ниже 0 1, но при этом для обработки экспериментальных результатов необходимо применять методы математической статистики.
В начале титрования средний отсчет измеряемой оптической плотности титруемого раствора полимерасоответствует нулевой точке. Затем обычно после нескольких дозированных добавлений осадителя оптическая плотность раствора ( D) начинает увеличиваться. Очень важно возможно точнее установить объем добавленного осадителя ( l / s), при котором показания правого барабана ( красная шкала) начинают изменяться.
Выбор кюветы - определяется оптимальным диапазоном измеряемых оптических плотностей
44. Способы определения концентрации веществ. Измерение высоких, низких оптических плотностей (дифференциальный метод). Анализ многокомпонентных систем.
1). Метод градуировочного графика.
2). Метод добавок.
1). Для определения концентрации вещества по методу градуировочного графика «ГГ» готовят серию стандартных растворов, концентрация которых охватывают интересующую нас область. Если данный раствор подчиняется основному закону поглощения, то для построения ГГ обычно находят 5 – 8 точек, отличающихся по концентрациям не менее чем на 30%.
Неизвестную концентрацию находят из градуировочного графика.
2). Метод добавок. Этот метод применим только при полном подчинении данного раствора основному закону светопоглощения. Сравнивают оптическую плотность исследуемого раствора и того же раствора с добавкой известного количества определяемого вещества.
Ax= εCxl
Ax+доб=ε(Cx+Cдоб)l
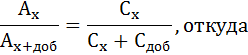

Методы абсорбционной спектрометрии применяют для анализа объектов окружающей среды, в основном, для определения металлов в воде, воздухе, почве, а также применяют в сельском хозяйстве, в медицине, в химической промышленности.
3.5. Дифференциальные фотометрические методы анализа
Дифференциальный метод применяют для повышения прецизионности результатов анализа при определении больших количеств веществ, а также для устранения мешающего влияния посторонних компонентов и исключения поглощения реактива.
Сущность дифференциального метода состоит в том, что оптические плотности исследуемого и стандартного окрашенных растворов измеряются не относительно чистого растворителя с нулевым поглощением, а по отношению к окрашенному раствору определяемого вещества с концентрацией СО, близкой к концентрации исследуемого раствора.
Дифференциальный метод в зависимости от способов измерения относительной оптической плотности исследуемого раствора и расчета его концентрации может иметь несколько вариантов. Наиболее часто используют вариант, в котором концентрация раствора сравнения меньше концентрации исследуемого раствора (СО < СХ). Измеренная экспериментально относительная оптическая плотность A представляет собой разность оптических плотностей фотометрируемого раствора и раствора сравнения.
AХ = АХ – АО = ?(СХ – СО)l, (3.15)
AСТ = АСТ – АО = ?(ССТ – СО)l. (3.16)
Концентрацию исследуемого раствора определяют либо при помощи градуировочной кривой, либо расчетным методом.
Для построения градуировочной кривой приготавливают серию стандартных растворов с концентрациями С1, С2, С3, ..., Сi, ..., Сn. (Сn > Сi… > С3 > С2 > С1) и измеряют их оптические плотности по отношению к окрашенному раствору сравнения с концентрацией СО.
По полученным данным строят градуировочную кривую (3.15), принимая за начало отсчета концентрацию раствора сравнения СО.
Измерив оптическую плотность исследуемого раствора AХ, по градуировочному графику определяют его неизвестную концентрацию СХ.
При расчетном способе определения концентрации исследуемого раствора учитывают, что отношение значений относительных оптических плотностей исследуемого и стандартного растворов соответствует отношению разности между концентрациями этих растворов и раствора сравнения:
AХ/АСТ = (СХ – СО)/(ССТ – СО), (3.17)
откуда
СХ = СО + AХ (ССТ – СО)/АСТ (3.18)
или
СХ = СО + F AХ. (3.19)
Отношение разности концентраций стандартного раствора и раствора сравнения к относительной оптической плотности стандартного раствора (ССТ – СО)/АСТ называют фактором пересчета F. Фактор пересчета – это обратный угловой коэффициент градуировочного графика. В одной серии измерений для определенного интервала концентраций исследуемого раствора F является постоянной величиной.
Данный вариант фотометрии широко применяют для экспрессного определения содержания основных компонентов с прецизионностью, не уступающей классическим методам анализа.
Физико-химический анализ — комплекс методов анализа физико-химических систем путем построения и геометрического анализа диаграмм состояния и диаграмм состав-свойство. Этот метод позволяет обнаружить существование соединений (например, медистого золота CuAu), существование которых невозможно подтвердить другими методами анализа. Первоначально исследования в области физико-химического анализа были сосредоточены на изучении зависимостей температур фазовых переходов от состава. Однако на рубеже XIX—XX веков Н. С. Курнаков показал, что любое физическое свойство системы является функцией состава, а для изучения фазового состояния можно использовать электропроводность, вязкость, поверхностное натяжение, теплоёмкость, коэффициент рефракции, упругость и другие физические свойства[1].
В основе теории физико-химического анализа лежат сформулированные Н. С. Курнаковым принципы соответствия и непрерывности. Принцип непрерывности утверждает, что если в системе не образуются новые фазы или не исчезают существующие, то при непрерывном изменении параметров системы свойства отдельныхфаз и свойства системы в целом изменяются непрерывно. Принцип соответствия утверждает, что каждому комплексу фаз соответствует определённый геометрический образ на диаграмме состав-свойство.
45. Фотометрические аналитические реагенты, требования к ним. Примеры практического применения метода.
Фотометрические методы
Эти методы занимают, пожалуй, исключительное место в работах химиков-аналитиков: им уделяется очень много внимания, число публикаций весьма велико. Преимущественно разработаны фотометрические методы определения элементов с использованием органических реагентов; многое сделано и по общим вопросам фотометрии.
Эти методы состоят в измерении поглощения лучистой энергии растворами анализируемых веществ. Характер спектра поглощения служит качественным признаком определяемого соединения, а величина поглощения выступает как количественная характеристика, позволяющая судить о содержании интересующего нас компонента. Дело в том, что поглощение лучистой энергии при прочих равных условиях пропорционально концентрации поглощающего вещества.
Понятие фотометрические охватывает приемы, которые именуются или именовались колориметрическими, фотоэлектроколориметрическими, спектрофотометрическими и собственно фотометрическими. Происхождение этих терминов связано со способом измерения светопоглощения и типом применяемого для этой цели прибора. Однако в основе всех способов лежит поглощение света определяемым веществом в видимой, ультрафиолетовой и инфракрасной области спектра.
Считают, что одним из первых фотометрические методы применил русский минералог и химик В.М. Севергин, работавший в конце ХVIII – начале ХIX веков. Во второй половине ХIХ века некоторые методы этого типа использовали на заводах, например в Нижнем Тагиле. Широкое же распространение фотометрия получила в Советском Союзе, начиная с 30-х годов ХХ века. По числу научных публикаций фотометрический метод занимает первое место. В очень большом масштабе метод используют в практике анализа самых разнообразных объектов. Достоинство этих приемов в довольно низком пределе обнаружения, доступности и простоте, сочетающихся во многих случаях с достаточной избирательностью и быстротой; точность определений также для ряда целей вполне удовлетворительна: относительная ошибка обычно составляет 5-10 %. Важно и то, что фотометрические методы разработаны практически для всех элементов и очень многих органических соединений.
В качестве фотометрических реагентов используют вещества различных классов. Из неорганических соединений – это галогениды и роданиды, перекись водорода, аммиак, соединения, дающие гетерополикислоты. Из более многочисленных органических реагентов можно назвать реагенты, содержащие в определенном сочетании гидроксильную и карбоксильную группу, в частности оксиазосоединения; реагенты с тиольной и тионной группами. Очень часто хороший аналитический эффект дают многокомпонентные соединения, например комплексы со смешанной координационной сферой.
В ряде аналитических центров синтезируются и испытываются новые фотометрические реагенты.
Чаще всего фотометрические методы используют для определения малых концентраций веществ. Однако можно определять и большие количества, если использовать так называемую дифференциальную фотометрию.
Органические фотометрические реагенты (ОФР)
Органические реагенты (ОР) наиболее широко применяются в спектрофотометрии в неорганическом анализе.
Чувствительные фотометрические методы, как правило, основаны на образовании двойных, тройных и даже четверных комплексов, хелатов или ионных пар металлов или неметаллов, особенно с окрашенными Органическими Реагентами.
Взаимодействуя с ионами металлов, большинство ОФР образуют или продукты, поглощающие свет в видимой или ультрафиолетовой области, или флуоресцирующие продукты.
ОФР относятся к одному из следующих классов:
1) органические реагенты (ОР), образующие хелатные комплексы (хелаты);
2) ОР, образующие ион-ассоциированные комплексы;
3) ОР, окисляющиеся или восстанавливающиеся ионами металлов и превращающиеся при этом в светопоглощающие или флуоресцирующие продукты, не содержащие металла.
Реакции первых двух классов являются наиболее важными.
Для фотометрических методов анализа (ФМА) пригодны только те ОР, при взаимодействии ионов металлов с которыми продукты реакции будут: или
а) значительно отличаться по фотометрическим свойствам (спектрам поглощения, флуоресценции) от этих же свойств избытка ОР, или
b) оптические свойства продукта реакции и ОР будут во многом близки, но реагент и продукт реакции легко разделяются.
Органические реагенты подразделяют на специфические, избирательные и групповые. К избирательным реагентам относят такие, которые реагируют лишь с определенной группой металлов или образуемые комплексы которых с различными металлами отличаются особыми физическими или химическими свойствами. Избирательный реагент называют специфическим, если он взаимодействует только с одним ионом (элементом). Подавляющее большинство органических реагентов является групповыми, которые взаимодействуют со многими элементами периодической системы.
Среди важнейших групповых органических реагентов, которые получили широкое распространение для определения индивидуальных элементов, следует назвать такие, как дитизон, диэтилдитиокарбаминат натрия и свинца, сульфохлорфенол С, диантипирилметан и некоторые другие.
Дата добавления: 2015-01-29; просмотров: 2817; Мы поможем в написании вашей работы!; Нарушение авторских прав |