КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Почему и как зависит скорость гомогенной химической реакции от температуры и концентрации реагирующих веществ?
Какие состояния компонентов принимают в качестве стандартных и как пересчитать компонент активности компонента раствора при переходе от одного стандартного состояния к другому? (136-144) Шварцман
Для растворов разных типов удобно выбирать и различные состояния в качестве стандартных.
Стандартное состояние следует выбирать так, чтобы в условиях, когда поведение реального раствора становится таким же, как идеального, активность совпадала бы с концентрацией и сохранилась бы форма уравнений, характеризующих тд свойства идеальных растворов.
Для идеальных растворов
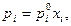 (Закон Рауля) (1)
(Закон Рауля) (1)
где 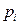 - парциальное давление пара i – го компонента над раствором;
- парциальное давление пара i – го компонента над раствором;
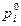 - парциальное давление пара i – го компонента над чистым компонентом.
- парциальное давление пара i – го компонента над чистым компонентом.
Активность i – го компонента в растворе определяется как отношение давления пара этого компонента над раствором к давлению его пара в стандартном состоянии 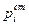 :
:
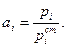 (2)
(2)
Сопоставление соотношений (1) и (2) показывает, что активность может совпадать с мольной долей в случае 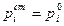 . Таким образом, для растворов, близких к идеальным, в качестве стандартного состояния принимается чистый компонент, и в этом случае
. Таким образом, для растворов, близких к идеальным, в качестве стандартного состояния принимается чистый компонент, и в этом случае
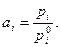 (3)
(3)
Отсюда ясно, что активность компонента в растворах, близких к совершенным, всегда меньше единицы, и только для компонента, находящегося в чистом состоянии,  .
.
Из уравнения (3) и из закона Рауля ( 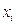
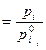 ) следует, что для совершенного раствора активность равна молярной доле. Если поведение раствора отклоняется от совершенного, то
) следует, что для совершенного раствора активность равна молярной доле. Если поведение раствора отклоняется от совершенного, то  . Степень этого отклонения определяется величиной коэффициента активности
. Степень этого отклонения определяется величиной коэффициента активности
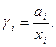 (4)
(4)
В случае совершенных растворов 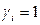 и
и 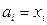 . Для реальных растворов коэффициент активности может быть как меньше единицы (отрицательные отклонения), так и больше (положительные отклонения).
. Для реальных растворов коэффициент активности может быть как меньше единицы (отрицательные отклонения), так и больше (положительные отклонения).
Из сказанного следует, что введение новой функции активности является формальным приемом, упрощающим термодинамические соотношения и позволяющим проводить расчеты равновесий с участием реальных растворов. Понятие активности само по себе не вскрывает механизма межчастичного взаимодействия в растворах, его недостаточно для полного описания системы, которое возможно лишь на основе молекулярно- статистических представлений. Однако измерения активности как для решения конкретных задач, так и для систематизации опытных данных, необходимы для развития теории растворов. Измерения активности дают возможность оценить отклонения в поведении реальных растворов от идеальных.
В разбавленных растворах для растворителя выполняется закон Рауля, а для растворенного вещества закон Генри: 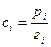 .
.
В случае растворов, близких к разбавленным, для растворителя в качестве стандартного состояния целесообразно принять этот компонент в чистом состоянии. При этом, как и для растворов, близким к совершенным, закон Рауля для растворителя может быть распространен на концентрированные растворы при условии замены молярной доли активностью, т.е. 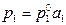 .
.
Для растворенного вещества за стандартное состояние принимают его гипотетическое (условное) состояние в растворе при концентрации, равной единице.
Для растворенного вещества выбор стандартного состояния должен быть сделан таким образом, чтобы в растворе любой концентрации соблюдался закон Генри. Сопоставление уравнения 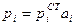 с уравнением показывает, что для этого необходимо, чтобы
с уравнением показывает, что для этого необходимо, чтобы 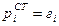 , т.е. давление пара растворенного вещества в стандартном состоянии должно быть численно равно величине коэффициента Генри (
, т.е. давление пара растворенного вещества в стандартном состоянии должно быть численно равно величине коэффициента Генри ( 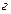 ). При этом
). При этом
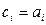 . (5)
. (5)
В разбавленном растворе 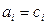 , а при любых более высоких концентрациях закон Генри сохраняется при замене концентрации активностью. При рассмотрении таких растворов также вводится коэффициент активности, равный отношению
, а при любых более высоких концентрациях закон Генри сохраняется при замене концентрации активностью. При рассмотрении таких растворов также вводится коэффициент активности, равный отношению 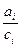 . В отличии от растворов, близких к совершенным, его принято обозначать буквой
. В отличии от растворов, близких к совершенным, его принято обозначать буквой 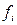 , т.е.
, т.е. 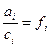 .
.
Таким образом, коэффициент активности компонента в растворе зависит как от способа выражения концентрации, так и от выбора стандартного состояния.
Пересчет активностей с одного стандартного состояния на другое осуществляется из того условия, что значение химического потенциала не зависит от выбора этих стандартных состояний и от способа выражения концентраций. (ТМП Сотников, Попель, Бороненков, с. 128-130)
Различают положительные и отрицательные уклонения свойств реальных растворов от идеальных. В чём причины таких уклонений с молекулярной точки зрения? При каких уклонениях и в каком случае возможно расслоение растворов? (124-133) Шварцман
Совершенные растворы характеризуются следующими свойствами: образование раствора не сопровождается тепловым эффектом (∆H=0), а изменение энтропии определяется уравнением (V.30 ∆S = -R [xlnx + (1-x)ln(1-x)].
Реальные растворы, вообще говоря, не обладают этими свойствами и для них не соблюдается закон Рауля. На рис. V.8 и V.9 представлены зависимости давления пара от состава для двух реальных растворов. Прямые штриховые линии соответствуют давлению пара, которое наблюдалось, если бы растворы были совершенными. На рис. V.8 показаны положительные отклонения, при которых давление пара выше, чем для совершенного раствора такой же концентрации, а на рис. V.9 — отрицательные отклонения, где давление пара меньше, чем для совершенного раствора
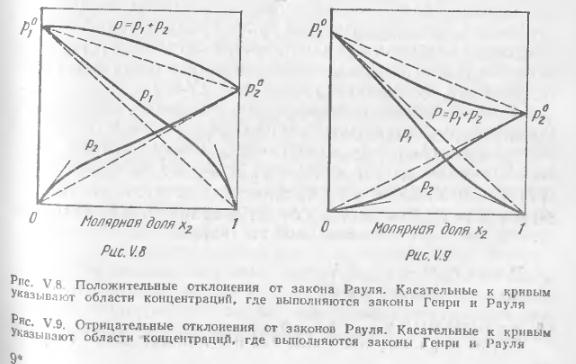
Наличие положительных отклонений показывает, что образование раствора затруднено по сравнению с совершенным. При больших положительных отклонениях жид. кости расслаиваются. Отрицательные отклонения указывают па более сильную тенденцию к образованию раствора, чем в случае совершенного раствора. Это означает, что имеются дополнительные причины, облегчающие образование раствора. Так как изменение энергии Гиббса выражается уравнением ∆G=∆H—T∆S, то отклонения в поведении реальных растворов от совершенных могут определяться как тем, что ∆H≠0, так и тем, что изменение энтропии при смешении не подчиняется уравнению
∆Sсов = -R [xlnx + (1-x)ln(1-x)].
Часто отклонения в поведении реальных растворов от идеальных законов обусловлены наличием теплоты смешения ∆H≠0. В этом случае при положительных отклонениях имеет место поглощение тепла при образовании раствора, а при отрицательных — выделение тепла. Для описания термодинамических свойств реальных растворов используют различные приближенные теории. Согласно одной из них — теории регулярных растворов — отличие от свойств совершенных растворов обусловлено наличием теплоты смешения, т. е. тем, что ∆H≠0. В то же время принимается, что изменение энтропии при образовании регулярного раствора остается таким же как у совершенного раствора идентичного состава и описывается уравнением (V.30).
Энтальпия образования бинарного регулярного раствора определяется выражением ∆H=Cx (1-x), где С—постоянная. Таким образом, с учетом уравнения (V.30) изменение энергии Гиббса при образовании регулярного раствора
∆G=∆H—T∆S = Cx (1-x) + RT [xlnx + (1-x)ln(1-x)] (V.34)
Если С>0, т. е. раствор образуется с поглощением тепла, то величина AG более положительна, чем при образовании совершенного раствора той же концентрации. Это означает, что тенденция к самопроизвольному образований раствора уменьшается по сравнению с совершенным и отклонения от закона Рауля положительны.
В системах, где С>0 и достаточно велика, первый член правой части уравнения (V.34) может по абсолютной величине превысить значение второго члена так, что ∆G>0. В этом случае раствор не может образовываться. Возможно, однако, что он существует при больших Т, но при понижении температуры он будет расслаиваться. При С<О образование раствора сопровождается выделением тепла и отклонения от закона Рауля отрицательны.
Растворы металлов, особенно переходных, не описываются теорией регулярных растворов. В частности, в бинарных сплавах не наблюдается концентрационная симметричность термодинамических свойств (∆G, ∆Н и ∆S), которая вытекает из уравнения (V.34).
15. Почему переход компонента в раствор сопровождается тепловым эффектом? Чем определяется знак теплового эффекта? В чём отличие дифференциальной теплоты растворения от интегральной, и как оно изменяется с изменением состава раствора? (см. лекции Ватолин 48)
При образовании раствора, когда в чистый растворитель вводится растворяемое вещество, появляется новая структура с иным расположением частиц, чем в чистом растворителе, которое будет зависеть от состава раствора. Изменяются и силы межмолекулярного взаимодействия. В растворе кроме взаимодействия между однородными молекулами появляется взаимодействие и между разнородными частицами. Частицы растворенного вещества взаимодействуют в растворе друг с другом и с молекулами растворителя.
Образование жидких растворов обычно сопровождается процессом сольватации. Под сольватацией понимают совокупность энергетических и структурных изменений, происходящих в растворе при взаимодействии частиц растворенного вещества с молекулами растворителя.
В действительности если взять бинарный раствор из компонентов А и В, то энергии парных связей 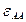 ,
,  ,
, 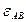 неодинаковы; смешение частиц сопровождается изменением внутренней энергии системы (
неодинаковы; смешение частиц сопровождается изменением внутренней энергии системы (  ). Неизбежно и изменение объема (
). Неизбежно и изменение объема ( 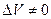 ) вследствие различия размеров частиц и возможной их деформации, поэтому
) вследствие различия размеров частиц и возможной их деформации, поэтому 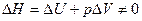 . Таким образом, образование раствора сопровождается тепловым эффектом (
. Таким образом, образование раствора сопровождается тепловым эффектом ( 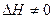 ).
).
Изменение энтальпии при переходе твердого, жидкого или газообразного вещества в раствор называют теплотой или энтальпией растворения. Эндотермические процессы характеризуются положительным изменением энтальпии ( 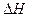 ), а экзотермические – отрицательным. Различают интегральные и дифференциальные теплоты растворения.
), а экзотермические – отрицательным. Различают интегральные и дифференциальные теплоты растворения.
Энтальпия раствора, состоящего из 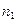 молей растворителя и
молей растворителя и 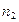 молей растворенного вещества, определяется уравнением
молей растворенного вещества, определяется уравнением
 (1)
(1)
Энтальпия исходной системы равна
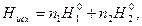 (2)
(2)
где 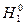 и
и  – молярные энтальпии чистых веществ.
– молярные энтальпии чистых веществ.
Тогда изменение энтальпии при образовании раствора равно
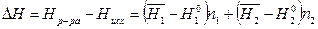 . (3)
. (3)
Интегральной теплотой растворения называют изменение энтальпии при растворении 1 моля вещества в некотором количестве чистого растворителя.
Таким образом, интегральную теплоту растворения(  ) можно записать в следующем виде
) можно записать в следующем виде
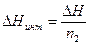 . (4)
. (4)
Интегральная теплота растворения зависит от концентрации полученного раствора и температуры. Так как можно получить растворы разной концентрации, то число интегральных теплот растворения может быть сколь угодно большим. Особый интерес представляют первая и полная интегральные теплоты растворения. Первой интегральной теплотой растворения называется изменение энтальпии при растворении одного моля вещества в бесконечно большом количестве чистого растворителя. В результате этого процесса образуется бесконечно разбавленный раствор. Полной интегральной теплотой растворения называется изменение энтальпии при растворении одного моля вещества в таком количестве чистого растворителя, которое необходимо для образования насыщенного раствора (с точки зрения термодинамики раствор называется насыщенным, когда химический потенциал чистого растворяемого вещества (твердого, жидкого, газообразного) равен химическому потенциалу этого вещества в растворе).
Дифференциальной или парциальной теплотой растворения называется изменение энтальпии при растворении одного моля вещества в бесконечно большом количестве раствора данной концентрации. В этом процессе концентрация раствора остается неизменной или, точнее, возрастает на бесконечно малую величину, которой можно пренебречь.
Энтальпия раствора, состоящего из молей растворителя и (  ) молей растворенного вещества, определяется уравнением
) молей растворенного вещества, определяется уравнением
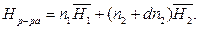 (5)
(5)
Энтальпия исходной системы равна
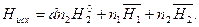 (6)
(6)
Тогда изменение энтальпии при образовании раствора равно
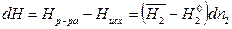 . (7)
. (7)
Таким образом, дифференциальная теплота растворения:
 (8)
(8)
Дифференциальная теплота растворения зависит от концентрации раствора. Дифференциальная теплота растворения одного моля вещества в бесконечно большом количестве насыщенного раствора называется последней теплотой растворения.
Интегральные теплоты растворения, кроме первой и полной, определяются опытным путем. Первая и полная интегральные теплоты растворения и все дифференциальные теплоты растворения находятся путем соответствующей обработки экспериментальных данных. Для этого строят график зависимости интегральной теплоты растворения ( )от числа молей растворенного вещества (n2) в одном и том же количестве чистого растворителя.
16. Какие режимы гетерогенной химической реакции Вам известны? Как и зачем выявляют эти режимы? Какие пути ускорения реакции Вы можете рекомендовать в различных режимах? (см. лекции Ватолин 96-102)
17. Почему поверхностные слои жидкостей обладают избыточной свободной энергией? Какие методы определения поверхностной энергии расплавов Вам известны? Каковы достоинства и недостатки важнейших из них в применении к расплавам? (см. лекции Ватолин 105-112)
18. Почему при равновесии состав поверхностного слоя отличается от состава глубинного раствора? Как определить различие? Как рассчитывают адсорбцию компонентов в бинарных и многокомпонентных растворах? (см. лекции Ватолин 112-117)
19. Э.Д.С. высокотемпературного гальванического элемента, в котором протекает реакция [Ni]тв+½O2газ=(NiO)ж равна 0,575 В при Т=1573 К и 0,536 В при Т=1673 К. Какие термодинамические характеристики реакции можно найти по приведённым данным?
В гальваническом элементе химического типа на каждом из электродов протекают процессы:
[N]i = (Ni2+) + 2e (1)
и
0,5O2газ + 2e = (O2-).(2)
Суммарное уравнение процесса выглядит:
[Ni]тв+½O2газ=(NiO)ж
Так как уравнение э.д.с. элемента определяется как разность равновесных потенциалов, то, согласно выражению
Е = φр(2) - φр(1) = φО2-0 – φNi0 + RT/2F*(Ln(PO20.5*a[Ni] / a(O2-)*a(Ni2+)))
С учетом изотермы реакции
Е = - RT / 2F [Ln(aNiO / P0.5O2*aNi) – Ln KNiO] = - 1 ΔGNiO/ 2F
В общем случае
Е = - (ΔG / zF)
где ΔG – изменение энергии Гиббса при образовании одного моля вещества в результате процесса, самопроизвольно протекающего при замыкании внешней цепи;
zF – количество электричества, прошедшее при этом через внешнюю цепь.
Это соотношение свидетельствует о том, что, измеряя Е, можно рассчитать величину ΔG и далее воспользоваться уравнением изотермы химической реакции для определения константы равновесия или активности одного из реагентов.
1) ΔG, Дж/моль NiO 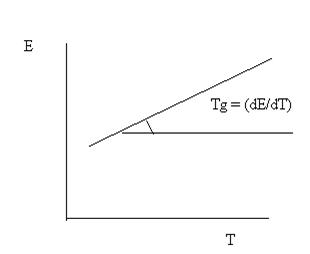
ΔG = - Е* zF
2) ΔS, Дж/моль*К
ΔS = - (dΔG/dT)
ΔS = zF(dE/dT)p
3) ΔH = - zFE + TzF(dE/dT)p,
так как ΔG = ΔH – Т* ΔS
4) Или константу равновесия реакции или активность одного из реагентов
- zFE = ΔG = RT[LnП - LnKp]
- zFE = RT[Ln(aNiO/aNi*P0.5O2)фак -LnKp]
Ni – чистый, aNi = 1
1)Kp по Шварцману –Темкину, тогда можно найти aNiO.
Любая хим. реакция для которой известна Kp
2) aNiO. знаем, тогда найдем Kp. Специально подбираем состав элемента, чтобы была известна aNiO
20. Вольтамперная характеристика электродного процесса (S2-)=[S]+2e имеет вид
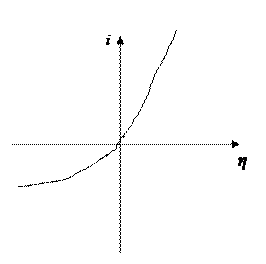
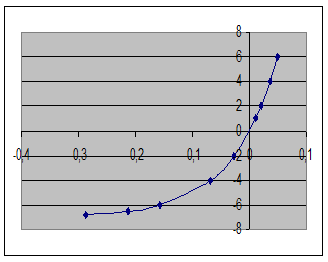
| i, mA | η, B |
| -2 -4 -6 -6,5 -6,8 | 0,0107 0,0203 0,0365 0,0500 0,0615 -0,0270 -0,0680 -0,1570 -0,2130 -0,2870 |
Какая стадия процесса лимитирует его скорость? Можно ли увеличить скорость десульфурации данного металла шлаком, уменьшая содержание FeO в шлаке?
А = В + ze
(S2-)=[S]+2e
Процесс перехода серы из шлака в металл, если в прямом направлении.
Данный процесс является диффузионным, вольт-амперная характеристика отражает, что замедлена диффузия вещества [S], а диффузия вещества (S2-) протекает легко и быстро.
in(A) → ∞
η = RT / zF*Ln(1 + i / in(B))
i = in(B)*(e zFη / RT – 1)
При η → - ∞, i → - in
А = В + ze
Процесс идет в обратном направлении, так как замедлена диффузия [S]/
i / in(B) = 1- СB(S) / СB(V),
СA(S) = СA(V)
Так как замедленной стадией является диффузия [S] в металлической фазе, и, как уже установлено, процесс (S2-)=[S]+2e протекает в обратном направлении, и если уменьшать FeO в шлаке, согласно реакции
(FeO) = [Fe] + [O]
Значит, суммируя эти реакции
[S]+2e = (S2-)
+
(Fe2+) - 2e = [Fe]
Следовательно, можно увеличить скорость десульфурации металла шлаком, уменьшая содержание FeO в шлаке.
Дата добавления: 2015-04-18; просмотров: 223; Мы поможем в написании вашей работы!; Нарушение авторских прав |