КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Масспектрометрия
Масс-спектрометр - прибор для разделения ионизированных молекул и атомов по их массам, основанный на воздействии магнитного или электрического поля на пучки ионов, летящих в вакууме. В масспектрометрах регистрация ионов осуществляется электрическими методами (сила тока), в масспектрографах – по потемнению фоточувствительного слоя. Схема прибора. Пример масс-спектра.

Рис.1. Схема масс-спектрометра. 1 – буферный резервуар, 2 – ионизационная камера, в которой поток молекул или атомов анализируемой пробы ионизируется с помозью потока ускоренных электронов, 3 – впуск образца, 4 – электронная пушка (источник потока укоренных электронов, с помощью которых ионизируются атомы и молекулы пробы), 5 – отрицательно заряженные пластины, с помощью которых ускоряется поток положительно заряженных ионов пробы, 6 – магнитное поле, 7 – траектория ионов с меньшей массой, 8 – траектория ионов с большей массой, 9 – к вакуумному насосу для создания вакуума на пути движения ионов, 10 – усилитель тока, 11 – устройство для записи силы тока в пике.
Масс-спектрометрия.[2], т.1, с.23 Этот экспериментальный метод применяется главным образом для двух целей: 1) определение относительных изотопных масс и изотопного содержания элементов; 2) определение относительных молекулярных масс и структуры органических соединений.
Как работает масс-спектрометр. Устройство масс-спектрометра схематически изображено на рис.1. Газообразный образец исследуемого вещества впускается в ионизационную камеру прибора. Здесь электронная пушка бомбардирует образец электронами, в результате чего образуются положительно заряженные ионы. Эти ионы преимущественно являются однозарядными. Затем ионы ускоряются электрическим полем и, попадая после этого в магнитное поле, отклоняются под его воздействием и движутся по искривленным круговым траекториям. Чем легче частицы, тем большее отклонение они испытывают. Интенсивность ионного пучка регистрируется электрическим устройством, в котором она усиливается и записывается.
Для получения масс-спектра постепенно повышают напряженность магнитного поля, в результате чего в детектор последовательно попадают ионы со все большим отношением массы к заряду. Для калибровки спектра обычно используют изотоп 12С, что позволяет непосредственно считывать со спектра относительные изотопные массы. Относительная интенсивность пиков масс-спектра определяет относительное изотопное содержание для исследуемого образца.
На рис.2 изображен масс-спектр природной смеси изотопов хлора. Из него видно, что относительное изотопное содержание 35CI в природной смеси равно 75%, а 37CI – 25%.
Принцип действия масс-спектрографа тот же, что и масс-спектрометра. Основное различие заключается в том, что для регистрации ионов в масс-спектрографе используется фотографическая пластинка, а не электрическое устройство.
Массовое число – это суммарное число протонов и нейтронов в атомном ядре.
Атомный номер – это число протонов в атомном ядре.
Изотопы химического элемента имеют одинаковый атомный номер, но разные массовые числа.
Относительная изотопная масса – это масса соответствующего нуклида, отнесенная к одной двенадцатой части массы ядра изотопа (нуклида) 12С.
Относительная атомная масса элемента – это средневзвешенное значение его относительных изотопных масс, вычисленное с учетом изотопного содержания данного элемента.
Относительные изотопные массы определяют с помощью масс-спектрометрии.

Рис. Масс-спектр природной смеси изотопов хлора, полученный с помощью масспектрометра. Указанное в периодической системе элементов усредненное массовое число природной смеси изотопов хлора можно получить из результатов вышеприведенного масс- спектра:
35· 0.75 + 37· 0.25 = 35.5 .
Существует еще радиоактивный изотоп хлора 36CI, полученный искусственным путем. В природной смеси его нет, следовательно, нет пика 36 в масс-спектре природной смеси. [9], том 1, с.23.
Активационный анализ
Параметры, характеризующие радиоактивный изотоп: массовое число, заряд ядра, вид радиоактивного распада (α, β, γ, n, К-захват и др.), период полураспада Т1/2. Связь периода полураспада Т1/2 с постоянной распада λ. Для радиоактивного изотопа углерода  массовое число 14, заряд ядра 6, вид распада β, период полураспада 5720 лет. Связь периода полураспада с постоянной распада Т1/2 = (In2)/λ. [2]
массовое число 14, заряд ядра 6, вид распада β, период полураспада 5720 лет. Связь периода полураспада с постоянной распада Т1/2 = (In2)/λ. [2]
Нейтронно-активационный анализ (используется в основном для выявления примесей в чистых веществах). Принцип. Пример реакции получения радиоактивного изотопа 28AI в результате нейтронной активации природного стабильного изотопа 27AI
27AI + 1n = 28AI.
Метод изотопного разбавления. Область применения. Например, для определения доли выделенного из смеси чистого вещества. [2]
Активационный анализоснован на измерении энергии излучения и периодов полураспада радиоактивных атомов, образующихся в исследуемом веществе при облучении его ядерными частицами (нейтронами, протонами, α - частицами и др.) и γ - квантами. Образование радиоактивных изотопов происходит в результате ядерных реакций. Количественный анализ обусловлен тем, что активность образующегося радиоактивного изотопа пропорциональна числу ядер исходного вещества, участвующего в ядерной реакции. Обычно используют относительный активационный анализ, основанный на сравнении активностей исследуемого образца и образца, в котором содержание определяемых элементов точно известно.
Наиболее распространен нейтроноактивационный анализ, при котором исследуемое вещество облучают тепловыми нейтронами с энергией 0.025 эВ, способными активировать почти все химические элементы, начиная с Na; при этом пределы обнаружения составляют 10-6 -10-14 г.
Достоинства активационного анализа: высокая специфичность, возможность одновременного определения ряда примесей в одной навеске образца, отсутствие поправки контрольного опыта, т.к. все операции, в том числе травление образца для удаления поверхностных загрязнений, проводят после облучения. Недостатки: относительно малая доступность источников активирующих частиц и γ – квантов (ядерных реакторов, циклотронов, нейтронных генераторов, линейных ускорителей и т.п.), радиационная опасность.
Основные области применения активационного анализа: анализ чистых веществ, в том числе материалов, применяемых в радиоэлектронике, атомной энергетике, авиационной промышленности и др.; анализ геологических объектов, экологические исследования, медицина.
Радиоактивные вещества испускают частицы нескольких видов: электроны (е), позитроны (е+), альфа-частицы (α), нейтроны (n) и др. Испускание этих частиц обычно (хотя и не всегда) сопровождается испусканием гамма – квантов (γ). К другому типу радиоактивного распада относится спонтанный захват ядром электрона с К-оболочки. Этот процесс, известный как К-захват, вызывает характеристическое для элемента рентгеновское К-излучение.
Частицы и излучение, испускаемое различными ядрами, сильно разнятся по энергии. Они характеристичны для распадающегося изотопа и, следовательно, могут быть использованы для идентификации. Частота, с которой происходят атомные распады, характеризуется специфичным для изотопа периодом полураспада. Это время, в течение которого распадается половина имеющихся атомов. Для известных радиоактивных изотопов время полураспада составляет от миллионных долей секунды до миллионов лет. Для аналитических измерений пригодны изотопы с периодами полураспада от нескольких часов до нескольких тысяч лет. Ряд некоторых пригодных для аналитических целей радиоизотопов представлен в таблице.
| Изотоп | Тип распада | Период полураспада | Энергия радиации, МэВ | |
| частицы | γ - излучения | |||
| 3Н | β- | 12.26 года | 0.0186 | нет |
| 14С | β- | 5720 лет | 0.155 | нет |
| 22Na | β+ (90%) ЭЗ (10%) | 2.58 года | 0.545 нет | нет 1.27 |
| 32Р | β- | 14.3 сут. | 1.71 | нет |
| 35S | β- | 87 сут. | 0.167 | нет |
| 36CI | β- | 3.0 ∙105 лет | 0.714 | Нет |
28Al β- 2.240 мин 1.24
Примечание: ЭЗ - электронный захват внутреннего электрона ядром, например,
2211Na + е = 2210Ne
в отличие от β+ распада
2211Na = 2210Ne + β+
Детекторы радиации. [5], с.504.
Детекторы ионизирующей радиации: фотографические, сцинтилляционные счетчики, газовые ионизационные детекторы (счетчик Гейгера). Поясняющие схемы. [2]
Фотографический детектор. Используется в основном для получения представления о распределении радиоактивного вещества на поверхности твердых образцов с помощью так называемой авторадиографии.
Авторадиография – метод регистрации распределения радиоактивных веществ в объекте. Фотографическая пленка, чувствительная к радиоактивному излучению, накладывается на поверхность, экспонируется и проявляется. Места наибольшего почернения на проявленной пленке соответствуют локализации радиоактивных частиц на поверхности объекта. Используется в медицине, биологии.
Сцинтилляционные счетчики. Попадая в флуоресцирующую среду, частица или квант излучения вызывают вспышку видимого света. По числу таких вспышек (сцинтилляций) можно судить о количестве частиц или фотонов в потоке радиации.
Сцинтилляторы бывают твердые и жидкие. Из твердых наиболее распространен иодид натрия, активированный таллием NaI (Tl). Исторически первым сцинтиллятором был сульфид цинка ZnS.
В жидких сцинтилляторах активное вещество находится в растворенной форме. Многие органические вещества, растворенные в подходящих растворителях, ведут себя как сцинтилляторы, например, антрацен (три бензольных кольца), 1,4-дифенилбензол, 2,5-дифенилоксазол.
Газовые ионизационные детекторы. Радиоактивное излучение (кроме нейтронного) вызывает ионизацию атомов и молекул в материалах. Измерение степени ионизации в газах или в полупроводниках служит основой общего метода обнаружения радиоактивного распада.
Полупроводниковые детекторы. Хорошие пропорциональные детекторы изготавливают на основе кристаллов кремния Si и германия Ge.
Измерение нейтронных потоков. Поскольку нейтроны не имеют заряда, они не вызывают ионизации. Но их можно обнаружить с помощью счетчика, наполненного газообразным фторидом бора BF3. Нейтроны реагируют с ядрами 10B, образуя 7Li и α - частицы в результате реакции 10B(n,α)7Li. Образующиеся α – частицы регистрируются счетчиком обычным образом.
105B + 1оn =73Li + 42Не
Нейтронно-активационный анализ. Многие элементы приобретают радиоактивность после облучения потоком нейтронов (01n), протонов (11p), дейтронов (12 H) или α – частиц ( 24He2+ ). Особенно широко применяют нейтронную активацию. Мощным источником потока нейтронов является ядерный реактор.
При бомбардировке исследуемого образца тепловыми нейтронами, кинетическая энергия которых меньше 0.2 эВ, обычно ядро мишени захватывает нейтрон и образуется новое ядро. Его масса на единицу больше, а заряд прежний, т.е. образуется изотоп исходного элемента. Часто новое ядро нестабильно и самопроизвольно распадается с испусканием частицы или γ - кванта (или того и другого), иными словами, ядро радиоактивно. Полученные таким путем радиоактивные изотопы сильно различаются по периоду полураспада, и благодаря этой и некоторой другой информации, например, гамма - спектрам, можно идентифицировать радионуклиды.
При проведении нейтронно-активационного анализа образец помещают в ядерный реактор и облучают его потоком нейтронов, в результате чего все способные к возбуждению элементы пробы становятся радиоактивными.

Рис. Нейтронно-активационный анализ алюминиевого сплава. Кривая распада активированного сплава. [Юинг], c.517. Обратите внимание, что данные о частоте распада (активности) представлены в полулогарифмических координатах: по оси ординат отложены десятичные логарифмы активности (суммарная скорость распада всех присутствующих в образце радиоактивных ядер (Бк) в данный момент времени), а по оси абсцисс – время (час.). Масштаб оси времени таков, что распад радиоактивных ядер алюминия - 28 на графике не заметен. 28AI распадается практически полностью в первые полчаса (область графика, вплотную примыкающая при данном масштабе к оси ординат). Зато хорошо видны составляющие распада радиоактивных изотопов 24Na и 56Mn, характеризующие наличие примесей в сплаве - стабильных натрия 23Na и марганца 55Mn.
. Связь между скоростью изменения количества оставшихся к моменту времени t радиоактивных атомов N и активностью А образца в этот же момент времени:
А = 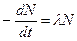 , N = N0 exp(-λt), InN = InN9 – λt,
, N = N0 exp(-λt), InN = InN9 – λt,
А = λ N = λN0 exp(-λt), А = А0 exp(-λt), InA = InAo – λt
Отсюда следует, что графики зависимости А от t в координатах InA – t для отдельных радиоизотопов (не смеси)будет прямолинейным.
Так называемая кривая распада отражает зависимость наведенной активности от времени. Эта кривая отражает активность всех радиоактивных элементов, образовавшихся в пробе после активации. Индивидуальные же радионуклиды можно идентифицировать, разлагая сложную кривую на составляющие (прямолинейные в полулогарифмических координатах). Период полураспада самого долгоживущего радионуклида можно определить по последнему участку кривой, в формирование которого менее стабильные компоненты уже не участвуют. Активность указанного радионуклида можно вычесть из общей кривой на участке, отвечающем меньшим временам. Затем так же можно определить следующий долгоживущий элемент, вычесть его вклад и т.д.
Проиллюстрируем этот способ на примере нейтронно-активационного анализа алюминиевого сплава. Небольшой его образец (массой около 25 мг) на 5 минут помещают в ядерный реактор. Затем регистрируют активность образца с помощью стандартного счетчика в течение 60 часов (рис.). В природе алюминий существует только в виде стабильного изотопа 27AI. После захвата нейтрона образуется 28AI, который подвергается β - распаду с образованием устойчивого 28Si. Поскольку образец состоял в основном из алюминия, излучение 28AI было очень мощным. Но поскольку период полураспада 28AI равен всего 2.3 минуты, через полчаса большинство ядер изотопа исчезло (на рисунке этот участок не заметен).
1327AI + 01n = 1328AI
1328AI = 1428Si + -10β
Из рисунка видно, что после примерно 40-часовой выдержки активность образца в полулогарифмических координатах линейно зависит от времени. Экстраполирование этой прямой линии влево дает для любого момента времени активность, обусловленную наличием элемента с периодом полураспада около 15 часов (значение определено из графика). По таблице изотопов этот элемент идентифицирован как 24Na с периодом полураспада 15.0 часов. На графике также имеется прямолинейный участок (от 1 до 18 часов), обусловленный присутствием изотопа с периодом полураспада примерно 2.5 ч - 56Mn (2.58 ч). Стабильный натрий и марганец, следовательно – примеси к алюминию.
Активация другими частицами. Активировать образец можно и бомбардировкой заряженными частицами – протонами 1Н+ , дейтронами 2Н+, тритонами 3Н+, ядрами гелия 3Не2+ и 4Не2+. Требуемую энергию (несколько десятков мегаэлектроновольт) сообщают частицам линейные ускорители, генераторы Ван-де-Граафа или циклотроны. При этом протекает такая же ядерная реакция, как и при облучении нейтронами: ядро-мишень захватывает частицу, образуя новое ядро с большей массой и зарядом. Новое ядро часто оказывается нестабильным и распадается, испуская частицы и излучение, или и то и другое вместе. Этот метод обладает высокой чувствительностью, особенно при обнаружении легких элементов (В, С, N, О), для которых предел обнаружения составляет порядка 10-7 %.
Общие вопросы анализа.
Чувствительность, предел обнаружения, динамический интервал. [5], c.541.
Чувствительность S аналитического метода или прибора определяется как отношение изменения аналитического сигнала R к изменению количества или концентрации C определяемого вещества:
S = dR/dC или S ≈ ΔR/ΔC
где ΔR и ΔC представляют собой малые, но конечные разности.
Наряду с чувствительностью часто используют параметр, называемый пределом обнаружения и определяемый как наименьшая концентрация, при которой аналитический сигнал становится настолько малым, что не распознается на фоне помех.
Максимальный размах концентраций, определяемых с известной надежностью, составляет область определяемых концентраций (динамический интервал). Ее нижняя граница определяется случайными флуктуациями (шумом) в сигнале прибора. Для не очень точного измерения отношение сигнал/шум (или сигнал/фон) должно быть равно, по крайней мере, двум, но для точных измерений оно должно быть больше. Верхняя граница определяемых концентраций связана с явлением «насыщения», например с растворимостью анализируемого соединения или возможностями детектора. Область определяемых концентраций для некоторых методов составляет 4 - 5 порядков измеряемой величины, для некоторых же ограничена одним порядком.
Воспроизводимость и правильность. [5], c.543.
Воспроизводимость, определяемая как разброс повторных определений, характеризуется стандартным отклонением (средним квадратичным отклонением Sn результата отдельного измерения хi от  ).. Чем меньше его значение, тем лучше воспроизводимость. Воспроизводимость тесно связана с правильностью, т.е. с близостью между полученным результатом и истинным значением. Результат с высокой воспроизводимостью может оказаться неправильным при наличии источников систематической ошибки, которая одинаково влияет на все параллельные измерения.
).. Чем меньше его значение, тем лучше воспроизводимость. Воспроизводимость тесно связана с правильностью, т.е. с близостью между полученным результатом и истинным значением. Результат с высокой воспроизводимостью может оказаться неправильным при наличии источников систематической ошибки, которая одинаково влияет на все параллельные измерения.
Воспроизводимость можно повысить, повторив анализ и проведя соответствующую обработку данных.
В любом методе титрования получают результаты серии измерений, по которым строят кривую титрования для определения точки эквивалентности. Проведение плавной кривой через точки улучшает общую воспроизводимость в такой же степени, как и получение того же числа повторных отсчетов без регистрации всей кривой титрования.
Сравнение со стандартными образцами. [5], c.544.
Большинство инструментальных аналитических методов основано на сравнении какого - либо физического свойства анализируемого вещества и стандарта или серии стандартов, содержащих то же вещество в известных количествах. Такое сравнение можно осуществить с помощью градуировочного графика, который представляет собой зависимость величины, выражающей это физическое свойство, от концентрации определяемого компонента. Иногда наклон кривой можно определить теоретически (из закона Бэра, уравнения Ильковича, уравнения Нернста и т.д.), и тогда удобнее рассчитывать концентрацию по уравнению, чем строить градуировочный график.
Другим общим приемом является сравнение анализируемого вещества с двумя стандартными образцами, один с несколько меньшим, а другой с несколько большим содержанием определяемого компонента, чем в анализируемом вещества. Преимущество такого метода заключается в том, что в таком небольшом интервале зависимость величины сигнала от концентрации можно считать линейной.
При всех сравнениях желательно, чтобы стандартные образцы и анализируемые вещества были как можно ближе друг к другу по составу. Это существенно снижает систематические ошибки, которые оказывают одинаковое влияние на все растворы.
Сравнение со стандартами не может улучшить воспроизводимость анализа, оно повышает его правильность.
Приготовление и хранение очень разбавленных растворов (от микро- до наномолярных) весьма затруднительно. Стенки сосудов, используемых для хранения, могут адсорбировать растворенные вещества, снижая тем самым концентрацию значительно ниже предполагаемого уровня. Иногда этого можно избежать, сполоснув сосуд раствором, который будет в нем храниться.
Стандартные добавки. [5], c.545
Нежелательное влияние основы (влияние посторонних веществ в пробе) на результат анализа можно уменьшить с помощью метода стандартных добавок, который заключается в добавлении стандарта непосредственно к пробе. Рассмотрим случай, когда после введения поправки на фон сигнал прибора R прямо пропорционален концентрации. Используя подстрочный индекс 0 для исходного раствора (пробы), запишем
 (1)
(1)
где концентрация представлена в виде отношения числа молей М к объему V. Добавим теперь порцию стандартного раствора, содержащую М1 молей в объеме V1. Запишем уравнение (1) для получившегося раствора
 (2)
(2)
Это выражение упрощается, если V1 будет намного меньше Vo, что легко сделать при использовании достаточно концентрированных стандартных растворов. Тогда
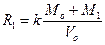 (3)
(3)
Исключив k/Vo в уравнении (3) с помощью уравнения (1) получим

 (4)
(4)
Этот прием исключает влияние большинства инструментальных переменных, представленных константой k.
Доказано, что наибольшая точность определения достигается, когда добавка М1 немного больше, чем количество определяемого вещества М0 :

Дата добавления: 2015-04-21; просмотров: 713; Мы поможем в написании вашей работы!; Нарушение авторских прав |