КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Статистическая обработка данных измерений
Три вида ошибок: систематические Δхсист, случайные Δхсл, грубые. Источники ошибок. Способы их уменьшения или устранения. Источники систематических ошибок: 1) методические (например, потери осадка вследствие его частичной растворимости); 2) инструментальные (например, систематическая ошибка взвешивания на аналитических весах составляет ± 0.0002 г.); 3) индивидуальные. [7], с.10.
Среднее результатов n параллельных измерений величины х при нормальном (гауссовском) распределении ошибок. 

Примечание. При распределении ошибок не нормальном вычисление среднего ведется по другим формулам.
Среднее квадратичное отклонение Sn результата отдельного измерения хi от  .. Дисперсия (разброс) σ2 результатов параллельных измерений при нормальном распределении.
.. Дисперсия (разброс) σ2 результатов параллельных измерений при нормальном распределении.
 , lim Sn2 = σ2
, lim Sn2 = σ2
n→∞
Среднее квадратичное отклонение среднего арифметического результатов n измерений

Выявление промахов, т.е. результатов отдельных измерений xi, не удовлетворяющих неравенству 
Доверительный интервал результатов параллельных измерений – интервал значений х, в котором истинное значение измеряемой величины находится с вероятностью β:

где 

где  определяется по значениям n и β из таблицы:
определяется по значениям n и β из таблицы:
| n | ||||||||||||
| β = 0.95 | 12.7 | 4.3 | 3.2 | 2.8 | 2.6 | 2.4 | 2.4 | 2.3 | 2.3 | 2.1 | 2.0 | |
| β = 0.99 | 63.7 | 9.9 | 5.8 | 4.6 | 4.0 | 3.7 | 3.5 | 3.4 | 3.3 | 2.9 | 2.8 |
Относительная погрешность δ среднего арифметического:
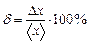
Погрешность Δy величины y, вычисляемой по формуле y = f(x1, x2, x3, … , xm), где xi - измеряемые величины, определяется по формуле  . Для каких значений х и Δх вычисляется функция Δу? Частные производные вычисляются в точке
. Для каких значений х и Δх вычисляется функция Δу? Частные производные вычисляются в точке 
Чувствительность определения. S = dR/dc, где … (не путать с Sn2)
Предел обнаружения компонента смеси. Динамический интервал.
Метрологические характеристики: чувствительность, предел обнаружения, воспроизводимость, точность, правильность результата измерений. [1], с. 6, 77.
Выявление систематических ошибок. Метод добавок или метод удвоения используют при отсутствии стандартных образцов и метрологически аттестованной методики анализа. Анализируют образец, используя оцениваемую методику. Затем удваивают массу анализируемой пробы или увеличивают (уменьшают) массу в иное число раз, снова находят содержание определяемого компонента в уже новой пробе и сравнивают результаты анализов. [1], с.53. [7], с.11.
Выявление систематических ошибок. Использование стандартных образцов. Общий состав стандартного образца должен быть близким к составу анализируемой пробы, а содержание определяемого компонента в стандартном образце должно быть точно известно. Анализ стандартного образца - наиболее надежный способ выявления наличия или отсутствия систематической ошибки и оценки правильности результата анализа.
Обработка данных измерений. При обработке данных измерений величин у и х, связанных зависимостью у =φ(x),желательно сначала представить зависимость у от х в координатах, в которых изучаемая зависимость будет прямолинейной: f(y) = c∙φ(x) + d. Затем можно использовать метод наименьших квадратов для нахождения параметров c и d этой прямолинейной зависимости. [11], с.354.Для примера приведем преобразование степенной y = a∙xb или экспоненциальной y = a∙ebx функции к линейному виду. [1], с.83. y = a∙xb, ln y = ln a + b∙ln x; y = a∙ebx, ln y = ln a + b∙x
Молекулярный спектральный анализ в УФ, видимой и ИК областях спектра.[7], с.305
Метод добавок стандарта. [7], с.340.
Метод применим, если выполняется основной закон светопоглощения.
Готовят два раствора: первый – анализируемый раствор с искомой концентрацией сх определяемого вещества и второй – анализируемый раствор, к которому прибавили точно известное количество (добавка стандарта) определяемого вещества, так что его концентрация во втором растворе равна сх + с, где с – точно известное увеличение концентрации за счет прибавления добавки стандарта.
Измеряют последовательно оптическую плотность D1 и D2 соответственно первого и второго растворов в одной и той же кювете при аналитической длине волны. С учетом выполнения основного закона светопоглощения можно записать
D1 = ε∙ сх ∙l
D2 = ε∙ [сх + c]∙l
откуда
 D1 ∙ сх + D1∙ с = D2∙ сх
D1 ∙ сх + D1∙ с = D2∙ сх

Хроматография. [8],c.667.
Термины: элюэнт – подвижная фаза. Сорбция – адсорбция и/или абсорбция. Этот же термин используется для обозначения ионообменных процессов.
Адсорбция (адсорбция газов поверхностью твердых тел). Атом или молекула газа, столкнувшись с поверхностью твердого тела, остается на ней в среднем в течение времени t, перед тем, как вернуться в газовую фазу. Для разных газов и на поверхностях разных твердых веществ это время «сидения» разное. Атомы и молекулы одних газов отражаются от поверхности сразу (t = 0). Эти газы не адсорбируются (не задерживаются) на поверхности данного твердого тела. Атомы или молекулы других газов характеризуются разным временем «сидения», иногда очень большим. В этом случае говорят об адсорбции данного газа поверхностью данного твердого тела. Газ концентрируется на поверхности адсорбента. Адсорбирующийся газ в этом случае называют адсорбатом, а твердое тело – адсорбентом. Процесс концентрирования адсорбата на поверхности адсорбента называют процессом адсорбции. Противоположный процесс - процесс покидания молекулами адсорбата поверхности адсорбента называют десорбцией. Процесс адсорбции, таким образом, является обратимым процессом.
Аналогичное рассуждение можно провести и для случая адсорбции атомов, молекул и ионов растворенных веществ на поверхности помещенного в жидкость твердого тела.
В практической деятельности в качестве адсорбентов используют, как правило, пористые твердые тела с сильно развитой внутренней поверхностью - активные угли, силикагели, активный оксид Al2O3, цеолиты. Величина адсорбции определяется количеством адсорбата, поглощенного единицей массы, объема или поверхности адсорбента. Эта величина зависит от температуры и давления, при которых происходит адсорбция, концентрации адсорбата в газовой фазе и пористости адсорбента (отношение объема пор к общему объему пористого вещества). Конечно, эта величина зависит и от конкретных веществ адсорбата и адсорбента.
Функция (или ее график), описывающая зависимость между величиной адсорбции и концентрацией адсорбата в газовой или жидкой фазе при достижении адсорбционного равновесия при постоянной температуре называется изотермой адсорбции.
В качестве примера приведем изотерму мономолекулярной адсорбции, теоретически выведенную Лэнгмюром:

где  - величина адсорбции адсорбата Х на поверхности адсорбента Y, (кг адсорбата Х)/(кг адсорбента Y);
- величина адсорбции адсорбата Х на поверхности адсорбента Y, (кг адсорбата Х)/(кг адсорбента Y);
 - величина адсорбции адсорбата Х на поверхности адсорбента Y при с(Х) →∞;
- величина адсорбции адсорбата Х на поверхности адсорбента Y при с(Х) →∞;
К – константа Лэнгмюра, зависящая от температуры, от конкретных веществ адсорбата Х и адсорбента Y, т.е., К = К(Х, Y, T).
с(Х) – концентрация адсорбата Х в газовой фазе (или в растворе), кг Х / м3.
Очевидно, что при с(Х) → 0 формула Лэнгмюра упрощается:

Аналогично выглядит уравнение Лэнгмюра и для случая адсорбции из жидкой фазы.
 |
Рис.1. График изотермы адсорбции Лэнгмюра в координатах а, с .

|
Рис. 2. График изотермы Лэнгмюра в координатах 1/а, 1/с .
Пусть теперь смесь газов просачивается через порошок адсорбента. В результате явления адсорбции начинается разделения смеси газов на ее составляющие. Как это происходит, смотри в разделе хроматография.
Хроматография - метод разделения веществ. Метод основан на различии в скоростях движения концентрационных зон исследуемых компонентов, которые перемещаются в потоке подвижной фазы (элюэнта) вдоль слоя неподвижной, причем исследуемые соединения распределены между обеими фазами. Обычно неподвижная фаза представляет собой сорбент с развитой поверхностью, а подвижная – поток газа (пара) или жидкости, фильтрующийся через слой сорбента. Различие в равновесном или кинетическом распределении компонентов между фазами – необходимое условие их хроматографического разделения.
В зависимости от агрегатного состояния подвижной фазы различают: 1) газовую хроматография, которую делят на газоадсорбционную хроматографию и газожидкостную хроматографию. 2) жидкостную хроматографию.
По геометрии сорбционного слоя неподвижной фазы различают колоночную и плоскостную хроматографию. К плоскостной относится тонкослойная хроматография и хроматография на бумаге. В колоночной обычно выделяют капиллярную хроматографию.
По механизму разделения различают ионообменную хроматографию, эксклюзионную хроматографию, осадочную хроматографию, аффинную хроматографию, адсорбционную и распределительную хроматографию. Последние два вида хроматографии основаны соответственно на различии сорбируемости разделяемых веществ адсорбентом и на различной растворимости их в элюэнте и в неподвижной фазе.
В зависимости от способа перемещения разделяемой смеси в колонке различают следующие виды хроматографии: проявительная, фронтальная и вытеснительная. В наиболее часто используемой проявительной хроматографии анализируемую смесь периодически вводят в поток подвижной фазы; в колонке смесь разделяется на отдельные компоненты, между которыми находятся зоны подвижной фазы. Во фронтальном варианте подвижная фаза с разделяемыми веществами непрерывно поступает в колонку; при этом только первый наименее сорбируемый компонент можно получить в чистом виде, вторая и последующие зоны содержат два и более компонентов. При вытеснительной хроматографии в колонку после разделяемой смеси вводят так называемый вытеснитель, который сорбируется лучше любого из анализируемых компонентов; это приводит к образованию примыкающих друг к другу зон разделяемых веществ. Во фронтальном и вытеснительном вариантах необходима регенерация колонки перед следующим опытом.
| Признак, по которому осуществляется классификация | ||
| По агрегатному состоянию подвижной фазы | газовая | газоадсорбционная |
| газожидкостная | ||
| Жидкостная | ||
| По геометрии сорбционного слоя | колоночная | капиллярная хроматография |
| плоскослойная | тонкослойная хроматография | |
| хроматография на бумаге | ||
| По механизму разделения | ионообменная хроматография | |
| эксклюзионная | ||
| осадочная | ||
| аффинная | ||
| адсорбционная | основана на различии сорбируемости разделяемых веществ адсорбентом | |
| распределительная | основана на различной растворимости разделяемых веществ в элюэнте и в неподвижной фазе. | |
| По способу перемещения разделяемой смеси по колонке | проявительная | |
| фронтальная | ||
| вытеснительная |
Хроматографию осуществляют с помощью приборов хроматографов, основные части которых – хроматографическая колонка и детектор (см. рис.1). 
Рис.1. Разделение смеси из трех компонентов 1, 2 и 3 на хроматографической колонке К с детектором Д: а – последовательные этапы разделения, б – хроматограмма. 1-й компонент несорбирующийся.

Рис.2. Газожидкостная хроматограмма смеси газов из десяти компонентов.1-инжекционный пик. В порядке увеличения времени удерживания: молекулярный водород Н2, молекулярный кислород О2, оксид углерода СО, метан СН4, этан С2Н6, углекислый газ СО2, этилен С2Н4, пропан С3Н8, ацетилен С2Н2, пропилен СН3СН=СН2. Инжекционный пик 1 (или момент времени 0) на этой хроматограмме соответствует моменту ввода образца в хроматограф. Время удерживания определяется химическим составом конкретного соединения и условиями эксперимента (температура колонки, скорость элюирования). [Фримантл.], т.1, стр.331.
В момент ввода пробы анализируемая смесь расположена в начале хроматографической колонки. Под действием потока подвижной фазы компоненты смеси начинают перемещаться вдоль колонки с различными скоростями; хорошо сорбируемые компоненты передвигаются вдоль слоя сорбента медленнее. Детектор на выходе из колонки автоматически непрерывно определяет концентрации определяемых соединений в подвижной фазе. Сигнал детектора регистрируется самописцем. Полученная диаграмма называется хроматограммой.
Дата добавления: 2015-04-21; просмотров: 340; Мы поможем в написании вашей работы!; Нарушение авторских прав |