КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Энтальпия образования химических соединений.
Энтальпией (теплотой) образования химического соединения DНТ называется изменение энтальпии в процессе получения одного моля этого соединения из простых веществ, устойчивых при данной температуре.
Стандартной энтальпией (теплотой) образования химического соединения DН  , обр называют изменение энтальпии в процессе образования одного моля этого соединения, находящегося в стандартном состоянии (Т = 298 К и r = 101,3 кПа), из простых веществ, также находящихся в стандартных состояниях и термодинамически устойчивых при данной температуре фазах и модификациях (таблица А.1).
, обр называют изменение энтальпии в процессе образования одного моля этого соединения, находящегося в стандартном состоянии (Т = 298 К и r = 101,3 кПа), из простых веществ, также находящихся в стандартных состояниях и термодинамически устойчивых при данной температуре фазах и модификациях (таблица А.1).
Стандартные энтальпии образования простых веществ принимаются равными нулю, если их агрегатные состояния и модификации устойчивы при стандартных условиях. Так, например, нулю равны стандартная теплота образования жидкого брома (а не газообразного) и графита (а не алмаза).
Стандартная энтальпия образования соединения – мера его термодинамической устойчивости, прочности, количественное выражение энергетических свойств соединения.
Термохимические расчеты.В основе большинства термохимических расчетов лежит следствие из закона Гесса: тепловой эффект химической реакции равен сумме теплот (энтальпий) образования продуктов реакции за вычетом суммы теплот (энтальпий) образования исходных веществ с учетом их стехиометрических коэффициентов в уравнении реакции.
DНх.р. = S DНобр. (прод. р-ции) - S DНобр (исх. в-в.) (9)
Уравнение (9) позволяет определять как тепловой эффект реакции по известным энтальпиям образования веществ, участвующих в реакции, так и одну из энтальпий образования, если известны тепловой эффект реакции и все остальные энтальпии образования.
Тепловой эффект химической реакции является энергетическим эффектом процесса, протекающего при постоянной температуре. Пользуясь справочными данными, которые относятся к 298 К, можно рассчитать тепловые эффекты реакций, протекающих при этой температуре. Однако при выполнении термохимических расчетов, допуская обычно незначительную ошибку, можно пользоваться стандартными значениями теплот образования даже тогда, когда условия протекания процесса отличаются от стандартных.
Тепловые эффекты фазовых превращений.Фазовые превращения часто сопутствуют химическим реакциям. Однако тепловые эффекты фазовых превращений, как правило, меньше тепловых эффектов химических реакций. Ниже приведены примеры термохимических уравнений некоторых фазовых превращений:
Н2О(ж) ® Н2О(г), DH = 44,0 кДж/моль,
Н2О(к) ® Н2О(ж), DH = 6,0 кДж/моль,
I2(к) ® I2(г), DH = 62,24 кДж/моль.
Исходя из выше приведенных данных можно отметить, что фазовый переход из более в менее конденсированное состояние ведет к повышению энтальпии системы (тепло поглощается – процесс эндотермический).
Т  Ж Г
Ж Г
Переход вещества из аморфного состояния в кристаллическое всегда сопровождается выделением теплоты (DH <0) – процесс экзотермический:
Sb(аморф) ® Sb(к), DH = -10,62 кДж/моль,
В2О3(аморф.) ® В2О3(к), DH = -25,08 кДж/моль.
Самопроизвольные и несамопроизвольные процессы.Многие процессы осуществляются самопроизвольно, т. е. без затраты работы извне. В результате их может быть получена работа против внешних сил, пропорциональная происшедшему изменению энергии системы. Так, самопроизвольно вода стекает по наклонному желобу или теплота передается от более нагретого тела к менее нагретому. В ходе самопроизвольного процесса система теряет способность производить полезную работу.
Самопроизвольный процесс не может протекать в обратном направлении так же самопроизвольно, как в прямом. Так, вода не может сама по себе перетекать вверх по наклонному желобу, а теплота не может сама по себе переходить от холодного тела к горячему. Чтобы перекачать воду наверх или передать теплоту от холодной части системы к горячей, необходимо совершить работу над системой. К процессам, обратным по отношению к самопроизвольным, применяется термин «несамопроизвольные».
При изучении химических взаимодействий очень важно оценить возможность или невозможность их самопроизвольного протекания при заданных условиях, выяснить химическое сродство веществ. Должен быть критерий, с помощью которого можно было бы установить принципиальную осуществимость, направление и пределы самопроизвольного течения реакции при тех или иных температурах и давлениях. Первый закон термодинамики такого критерия не дает. Тепловой эффект реакции не определяет направления процесса: самопроизвольно могут протекать как экзотермические, так и эндотермические реакции.
Критерий самопроизвольного протекания процесса в изолированных системах дает второй закон термодинамики. Прежде чем перейти к рассмотрению этого закона, введем представление о термодинамической функции состояния системы, называемой энтропией.
Энтропия.Для характеристики состояния некоторого количества вещества, являющегося совокупностью очень большого числа молекул, можно или указать температуру, давление и другие термодинамические параметры состояния системы, или указать мгновенные координаты каждой молекулы (xi, yi, zi) и скорости перемещения по всем трем направлениям (vxi,vyi,vzi). В первом случае охарактеризовывается макросостояние системы, во втором – микросостояние. Каждому макросостоянию отвечает огромное число микросостояний. Число микросостояний, с помощью которых осуществляется данное макросостояние, называют термодинамической вероятностью состояния системы и обозначают W.
Термодинамическая вероятность состояния системы, состоящей всего из 10 молекул газа, примерно 1000, а ведь только в 1 см3 газа содержится 2,7×1019 молекул (н. у.). Чтобы перейти к более удобным для восприятия и расчетов числам, в термодинамике используют не величину W, а ее логарифм lnW. Последнему можно придать размерность (Дж/К), умножив на константу Больцмана k:
klnW = S. (10)
Величину S называют энтропией системы.
Энтропия – термодинамическая функция состояния системы и ее величина зависит от количества рассматриваемого вещества. Поэтому целесообразно относить величину энтропии к одному молю вещества (Дж/(моль×К)) и выражать как
RlnW = S. (11)
где R = kNA – молярная газовая постоянная;
NA – постоянная Авогадро.
Из уравнения (11) следует, что энтропия системы увеличивается пропорционально логарифму термодинамической вероятности состояния W. Это соотношение лежит в основе современной статистической термодинамики.
При р = const энтропия является функцией температуры Т, причем температура замерзания и температура кипения – это те точки, в которых энтропия изменяется особенно резко, скачкообразно.
Итак, энтропия S является мерой неупорядоченности системы. «Носителями» энтропии являются газы. Если при реакции увеличивается число молей газообразных веществ, то увеличивается и энтропия. Т.е. не производя расчетов можно при необходимости определить знак изменения энтропии системы:
C(к) + О2(г) = СО2(г), DS » 0;
2C(к) + О2(г) = 2СО(г), DS > 0;
N2(г) + 3H2(г) = 2NH3(г), DS < 0.
В таблице А.1 приведены значения S некоторых веществ (обратите внимание на то, что известны абсолютные значения энтропии веществ, в то время как абсолютные значения функции U и H не известны).
Т.к. энтропия есть функция состояния системы, то изменение энтропии (DS) при химической реакции равно сумме энтропий продуктов реакции за вычетом суммы энтропий исходных веществ с учетом их стехиометрических коэффициентов в уравнении реакции.
DSх.р. = S Sобр. (прод. р-ции) - S Sобр (исх. в-в.) (12)
Направление и предел протекания процессов в изолированных системах. Второй закон термодинамики.Изолированные системы не обмениваются с внешней средой ни теплотой, ни работой. На основании уравнения (9) можно утверждать, что при q = 0 и A = 0 величина DU тоже равна нулю, т. е. внутренняя энергия изолированной системы постоянна (U=const); постоянен и ее объем (V = const). В изолированных системах самопроизвольно идут только те процессы, которые сопровождаются ростом энтропии системы: DS>0; при этом пределом самопроизвольного течения процесса является достижение максимальной для данных условий энтропии Smax.
Рассмотренное положение представляет одну из формулировок второго закона термодинамики (закон имеет статистический характер, т. е. применим лишь к системам, состоящим из очень большого числа частиц). Требование постоянства внутренней энергии и объема системы исключает использование энтропии как критерия направления и предела протекания химических реакций, при которых внутренняя энергия веществ неизбежно меняется, а также совершается работа расширения против внешнего давления.
Энтропийный и энтальпийный факторы химических реакций, протекающих в изобарно-изотермических условиях.Движущей силой процесса, протекающего в изобарно-изотермических условиях, может быть или стремление системы перейти в состояние с наименьшей энергией, т. е. выделить теплоту в окружающую среду, уменьшить энтальпию (DH<0), или стремление системы перейти в состояние с наибольшей термодинамической вероятностью, т. е. увеличить энтропию (DS>0). Если процесс протекает так, что DH=0, то рост энтропии становится его единственной движущей силой. И, наоборот, при условии DS = 0 единственной движущей силой процесса является убыль энтальпии. В связи с этим можно говорить об энтальпийном DH и энтропийном TDS факторах процесса.
Максимальная работа.Голландский физико-химик Вант-Гофф предложил новую теорию химического сродства, которая, не объясняя природы химического сродства, ограничивается указанием способа его измерения, т. е. дает количественную оценку химическому сродству.
Вант-Гофф использует в качестве меры химического сродства максимальную работу А 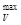 или А
или А  для реакций, протекающих при V, T = const или р, Т = const соответственно.
для реакций, протекающих при V, T = const или р, Т = const соответственно.
Максимальная работа равна энергии, которую нужно приложить к системе, чтобы остановить реакцию, т. е. преодолеть, силы химического сродства. Поскольку реакция протекает в направлении совершения положительной максимальной работы, знак А или А определяет направление самопроизвольного течения химического взаимодействия.
Максимальная работа при постоянном объеме равна
А = -DU + TDS (13)
или
А = -(U2 – U1) + T(S2 – S1) = -[(U2 – TS2) – (U1 – TS1)] (14)
где U1, S1 и U2, S2 – величина внутренней энергии и энтропии системы в исходном и конечном состояниях соответственно.
Разность (U - TS) называют энергией Гельмгольца системы и обозначают буквой F. Таким образом,
А = -DF. (15)
Дата добавления: 2015-04-16; просмотров: 1610; Мы поможем в написании вашей работы!; Нарушение авторских прав |