КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Хозяйствующие субъекты без образования юридического лица
Величина потенциала хлорсеребряного электрода зависит от активности ионов хлора; данная зависимость имеет следующий вид:
 (III.51)
(III.51)
Чаще всего в качестве электрода сравнения используется насыщенный хлорсеребряный электрод, потенциал которого зависит только от температуры.
Индикаторные электроды.
Электроды, обратимые относительно иона водорода, используются на практике для определения активности этих ионов в растворе (и, следовательно, рН раствора) потенциометрическим методом, основанном на определении потенциала электрода в растворе с неизвестным рН и последующим расчетом рН по уравнению Нернста. В качестве индикаторного электрода может использоваться и водородный электрод, однако работа с ним неудобна и на практике чаще применяются хингидронный и стеклянный электроды.
Хингидронный электрод, относящийся к классу окислительно-восстановительных электродов (см. ниже), представляет собой платиновую проволоку, опущенную в сосуд с исследуемым раствором, в который предварительно помещают избыточное количество хингидрона С6Н4О2·С6Н4(ОН)2 – соединения хинона С6Н4О2 и гидрохинона С6Н4(ОН)2, способных к взаимопревращению в равновесном окислительно-восстановительном процессе, в котором участвуют ионы водорода:
С6Н4О2 + 2Н+ + 2е- ––> С6Н4(ОН)2
Хингидронный электрод является т.н. окислительно-восстановительным электродом (см. разд. 3.5.5); зависимость его потенциала от активности ионов водорода имеет следующий вид:
 (III.52)
(III.52)
Стеклянный электрод, являющийся наиболее распространенным индикаторным электродом, относится к т.н. ионоселективным или мембранным электродам. В основе работы таких электродов лежат ионообменные реакции, протекающие на границах мембран с растворами электролитов; ионоселективные электроды могут быть обратимы как по катиону, так и по аниону.
Принцип действия мембранного электрода заключается в следующем. Мембрана, селективная по отношению к некоторому иону (т.е. способная обмениваться этим ионом с раствором), разделяет два раствора с различной активностью этого иона. Разность потенциалов, устанавливающаяся между двумя сторонами мембраны, измеряется с помощью двух электродов. При соответствующем составе и строении мембраны её потенциал зависит только от активности иона, по отношению к которому мембрана селективна, по обе стороны мембраны.
Наиболее часто употребляется стеклянный электрод в виде трубки, оканчивающейся тонкостенным стеклянным шариком. Шарик заполняется раствором НСl с определенной активностью ионов водорода; в раствор погружен вспомогательный электрод (обычно хлорсеребряный). Потенциал стеклянного электрода с водородной функцией (т.е. обратимого по отношению к иону Н+) выражается уравнением
 (III.53)
(III.53)
Необходимо отметить, что стандартный потенциал ε°ст для каждого электрода имеет свою величину, которая со временем изменяется; поэтому стеклянный электрод перед каждым измерением рН калибруется по стандартным буферным растворам с точно известным рН.
3.5.5 Окислительно-восстановительные электроды
В отличие от описанных электродных процессов в случае окислительно-восстановительных электродов процессы получения и отдачи электронов атомами или ионами происходят не на поверхности электрода, а только в растворе электролита. Если опустить платиновый (или другой инертный) электрод в раствор, содержащий двух- и трехзарядные ионы железа и соединить этот электрод проводником с другим электродом, то возможно либо восстановление ионов Fe3+ до Fe2+ за счет электронов, полученных от платины, либо окисление ионов Fe2+ до Fe3+ с передачей электронов платине. Сама платина в электродном процессе не участвуют, являясь лишь переносчиком электронов. Такой электрод, состоящий из инертного проводника первого рода, помещенного в раствор электролита, содержащего один элемент в различных степенях окисления, называется окислительно-восстановительным или редокс-электродом. Потенциал окислительно-восстановительного электрода также определяют относительно стандартного водородного электрода:
Pt, H2 / 2H+ // Fe3+, Fe2+ / Pt
Зависимость потенциала редокс-электрода εRO от концентрации (активности) окисленной [Ox] и восстановленной форм [Red] для окислительно-восстановительной реакции, в которой не участвуют никакие другие частицы, кроме окислителя и восстановителя, имеет следующий вид (здесь n – число электронов, участвующих в элементарном акте окислительно-восстановительной реакции):
 (III.54)
(III.54)
Из данного выражения следует уравнение для потенциала металлического электрода (III.40), т.к. активность атомов металла (восстановленной формы) в материале электрода равна единице.
В случае более сложных систем в выражении для окислительно-восстановительного потенциала фигурируют концентрации всех участвующих в реакции соединений, т.е. под окисленной формой следует понимать все соединения в левой части уравнения реакции
Ох + ne- ––> Red,
а под восстановленной – все соединения в правой части уравнения. Так, для окислительно-восстановительных реакций, протекающих с участием ионов водорода
Ох + ne- + mH+ ––> Red,
уравнение Нернста будет записываться следующим образом:
 (III.55)
(III.55)
При составлении гальванических элементов с участием редокс-электрода электродная реакции на последнем в зависимости от природы второго электрода может быть либо окислительной, либо восстановительной. Например, если составить гальванический элемент из электрода Pt / Fe3+, Fe2+ и второго электрода, имеющего более положительный электродный потенциал, то при работе элемента редокс-электрод будет являться анодом, т.е. на нем будет протекать процесс окисления:
Fe2+ ––> Fe3+ + e-
Если потенциал второго электрода будет меньше, чем потенциал электрода Pt / Fe3+, Fe2+, то на последнем будет протекать реакция восстановления и он будет являться катодом:
Fe3+ + e- ––> Fe2+
Знание величин электродных потенциалов позволяет определить возможность и направление самопроизвольного протекания любой окислительно-восстановительной реакции при одновременном наличии в растворе двух или более окислительно-восстановительных пар. Восстановленная форма любого элемента или иона будет восстанавливать окисленную форму другого элемента или иона, имеющего более положительный электродный потенциал.
4.1 ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ И АДСОРБЦИЯ
4.1.1 Поверхностная энергия. Адсорбция
До сих пор свойства гетерогенных систем описывались с помощью параметров и функций состояния, характеризующих каждую из фаз в целом. Однако свойства участка фазы, примыкающего к её поверхности, отличаются от свойств фазы в объеме: фактически частицы, находящиеся на поверхности каждой фазы, образуют особую поверхностную фазу, свойства которой существенно отличаются от свойств внутренних областей фазы. Частицы, расположенные на поверхности, находятся в другом окружении по сравнению с частицами, находящимися в объеме фазы, т.е. взаимодействуют как с однородными частицами, так и с частицами другого рода. Следствием этого является то, что средняя энергия gs частицы, находящейся на поверхности раздела фаз, отличается от средней энергии такой же частицы в объеме фазы gv (причем энергия частицы на поверхности может быть как больше, так и меньше энергии частицы в объеме). Поэтому важнейшей характеристикой поверхностной фазы является поверхностная энергия Gs – разность средней энергии частицы, находящейся на поверхности, и частицы, находящейся в объеме фазы, умноженная на число частиц на поверхности N:
 (IV.1)
(IV.1)
 (IV.2)
(IV.2)
Очевидно, что общая величина поверхностной энергии фазы будет определяться величиной её поверхности S. Поэтому для характеристики поверхности раздела, отделяющей данную фазу от другой, вводится понятие поверхностное натяжение σ – отношение поверхностной энергии к площади поверхности раздела фаз; величина поверхностного натяжения зависит только от природы обеих фаз. Как и поверхностная энергия фазы, поверхностное натяжение может иметь как положительное, так и отрицательное значение. Поверхностное натяжение положительно, если находящиеся на поверхности частицы взаимодействуют с частицами этой же фазы сильнее, чем с частицами другой фазы (и, следовательно, gs > gv). Согласно принципу минимума свободной энергии, любая фаза будет стремиться самопроизвольно уменьшить свою поверхностную энергию; поэтому в случае положительного поверхностного натяжения (σ > 0) фаза стремится уменьшить свою поверхность. В случае если σ < 0, поверхностная энергия фазы будет уменьшаться при увеличении площади поверхности.
Влияние поверхностного слоя фазы на её общие свойства определяется долей частиц, находящихся на поверхности, от общего числа составляющих данную фазу частиц, т.е. величиной удельной поверхности фазы S/V (поверхности, приходящейся на единицу объема). Свободную энергию фазы G можно представить как сумму поверхностной Gs и объемной Gv энергий, пропорциональных соответственно площади поверхности и объему фазы:
 (IV.3)
(IV.3)
Разделив это выражение на объем фазы, получаем:
 (IV.4)
(IV.4)
Из уравнения (IV.4) следует, что при одном и том же количестве фазы (т.е. неизменном объеме) вклад поверхностной энергии в общую энергию фазы возрастает с увеличением удельной поверхности или, иначе говоря, степени дисперсности (раздробленности) фазы. В случае, когда степень дисперсности фазы невелика (удельная поверхность незначительна), вкладом поверхностной энергии в полную энергию фазы обычно пренебрегают. Вклад поверхностного слоя в свойства фазы и системы в целом учитывают при изучении дисперсных систем – гетерогенных систем, одна из фаз которой является сплошной (дисперсионная среда), а другая – раздробленной (дисперсная фаза).
На границе конденсированной (т.е. твердой или жидкой) фазы с газом поверхностное натяжение всегда положительно, поскольку частицы конденсированной фазы взаимодействуют друг с другом сильнее, чем с молекулами газа. Согласно принципу минимума свободной энергии, конденсированная фаза будет стремиться самопроизвольно уменьшить свою поверхностную энергию. Это может быть результатом либо уменьшения площади поверхности фазы (именно поэтому капля жидкости в невесомости принимает форму сферы), либо уменьшения поверхностного натяжения при появлении на поверхности раздела фаз новых частиц – молекул газа либо растворенного вещества. Процесс самопроизвольного изменения концентрации какого-либо вещества у поверхности раздела двух фаз называется адсорбцией. Адсорбентом называется вещество, на поверхности которого происходит изменение концентрации другого вещества – адсорбата.
4.1.2 Адсорбция на границе раствор – пар
В жидких растворах поверхностное натяжение σ является функцией от концентрации растворенного вещества. На рис. 4.1 представлены три возможных зависимости поверхностного натяжения от концентрации раствора (т.н. изотермы поверхностного натяжения). Вещества, добавление которых к растворителю уменьшает поверхностное натяжение, называют поверхностно-активными (ПАВ), вещества, добавление которых увеличивает или не изменяет поверхностное натяжение – поверхностно-инактивными (ПИАВ).

Рис. 4.1 Изотермы поверхностного Рис. 4.2 Изотерма адсорбции натяжения растворов ПАВ (1, 2) и ПАВ на границе раствор – пар ПИАВ (3)
Уменьшение поверхностного натяжения и, следовательно, поверхностной энергии происходит в результате адсорбции ПАВ на поверхности раздела жидкость – пар, т.е. того, что концентрация поверхностно-активного вещества в поверхностном слое раствора оказывается больше, чем в глубине раствора.
Количественной мерой адсорбции на границе раствор-пар является поверхностный избыток Г (гамма), равный числу молей растворенного вещества в поверхностном слое. Количественное соотношение между адсорбцией (поверхностным избытком) растворенного вещества и изменением поверхностного натяжения раствора с ростом концентрации раствора определяет изотерма адсорбции Гиббса:
 (IV.5)
(IV.5)
График изотермы адсорбции ПАВ представлен на рис. 4.2. Из уравнения (IV.5) следует, что направление процесса – концентрирование вещества в поверхностном слое или, наоборот, нахождение его в объеме жидкой фазы – определяется знаком производной dσ/dС. Отрицательная величина данной производной соответствует накоплению вещества в поверхностном слое (Г > 0), положительная – меньшей концентрации вещества в поверхностном слое по сравнению с его концентрацией в объеме раствора.
Величину g = –dσ/dС называют также поверхностной активностью растворенного вещества. Поверхностную активность ПАВ при некоторой концентрации С1 определяют графически, проводя касательную к изотерме поверхностного натяжения в точке С = С1; при этом поверхностная активность численно равна тангенсу угла наклона касательной к оси концентраций:
 (IV.6)
(IV.6)
Нетрудно заметить, что с ростом концентрации поверхностная активность ПАВ уменьшается. Поэтому поверхностную активность вещества обычно определяют при бесконечно малой концентрации раствора; в этом случае её величина, обозначаемая gо, зависит только от природы ПАВ и растворителя. Исследуя поверхностное натяжение водных растворов органических веществ, Траубе и Дюкло установили для гомологических рядов поверхностно-активных веществ следующее эмпирическое правило:
В любом гомологическом ряду при малых концентрациях удлинение углеродной цепи на одну группу СН2 увеличивает поверхностную активность в 3 – 3.5 раза.
Для водных растворов жирных кислот зависимость поверхностного натяжения от концентрации описывается эмпирическим уравнением Шишковского:
 (IV.6a)
(IV.6a)
Здесь b и K – эмпирические постоянные, причём значение b одинаково для всего гомологического ряда, а величина К увеличивается для каждого последующего члена ряда в 3 – 3,5 раза.

Рис. 4.3 Предельная ориентация молекул ПАВ в поверхностном слое
Молекулы большинства ПАВ обладают дифильным строением, т.е. содержат как полярную группу, так и неполярный углеводородный радикал. Расположение таких молекул в поверхностном слое энергетически наиболее выгодно при условии ориентации молекул полярной группой к полярной фазе (полярной жидкости), а неполярной – к неполярной фазе (газу или неполярной жидкости). При малой концентрации раствора тепловое движение нарушает ориентацию молекул ПАВ; при повышении концентрации происходит насыщение адсорбционного слоя и на поверхности раздела фаз образуется слой "вертикально" ориентированных молекул ПАВ (рис. 4.3). Образование такого мономолекулярного слоя соответствует минимальной величине поверхностного натяжения раствора ПАВ и максимальному значению адсорбции Г (рис. 4.1-4.2); при дальнейшем увеличении концентрации ПАВ в растворе поверхностное натяжение и адсорбция не изменяются.
1.3 Адсорбция на границе твердое тело – газ
При адсорбции газов на твердых телах описание взаимодействия молекул адсорбата и адсорбента представляет собой весьма сложную задачу, поскольку характер их взаимодействия, определяющий характер адсорбции, может быть различным. Поэтому обычно задачу упрощают, рассматривая два крайних случая, когда адсорбция вызывается физическими или химическими силами – соответственно физическую и химическую адсорбцию.
Физическая адсорбция возникает за счет ван-дер-ваальсовых взаимодействий. Она характеризуется обратимостью и уменьшением адсорбции при повышении температуры, т.е. экзотермичностью, причем тепловой эффект физической адсорбции обычно близок к теплоте сжижения адсорбата (10 – 80 кДж/моль). Таковой является, например, адсорбция инертных газов на угле.
Химическая адсорбция (хемосорбция) осуществляется путем химического взаимодействия молекул адсорбента и адсорбата. Хемосорбция обычно необратима; химическая адсорбция, в отличие от физической, является локализованной, т.е. молекулы адсорбата не могут перемещаться по поверхности адсорбента. Так как хемосорбция является химическим процессом, требующим энергии активации порядка 40 – 120 кДж/моль, повышение температуры способствует её протеканию. Примером химической адсорбции является адсорбция кислорода на вольфраме или серебре при высоких температурах.
Следует подчеркнуть, что явления физической и химической адсорбции чётко различаются в очень редких случаях. Обычно осуществляются промежуточные варианты, когда основная масса адсорбированного вещества связывается сравнительно слабо и лишь небольшая часть – прочно. Например, кислород на металлах или водород на никеле при низких температурах адсорбируются по законам физической адсорбции, но при повышении температуры начинает протекать химическая адсорбция. При повышении температуры увеличение химической адсорбции с некоторой температуры начинает перекрывать падение физической адсорбции, поэтому температурная зависимость адсорбции в этом случае имеет четко выраженный минимум (рис. 4.4).

Рис. 4.4 Зависимость объема адсорбированного никелем водорода от температуры
При постоянной температуре количество адсорбированного вещества зависит только от равновесных давления либо концентрации адсорбата; уравнение, связывающее эти величины, называется изотермой адсорбции.
4.1.4 Теории адсорбции
Единой теории, которая достаточно корректно описывала бы все виды адсорбции на разных поверхностях раздела фаз, не имеется; рассмотрим поэтому некоторые наиболее распространенные теории адсорбции, описывающие отдельные виды адсорбции на поверхности раздела твердое тело – газ или твердое тело – раствор.
Теория мономолекулярной адсорбции Ленгмюра
Теория мономолекулярной адсорбции, которую разработал американский химик И. Ленгмюр, основывается на следующих положениях.
1) Адсорбция является локализованной и вызывается силами, близкими к химическим.
2) Адсорбция происходит не на всей поверхности адсорбента, а на активных центрах, которыми являются выступы либо впадины на поверхности адсорбента, характеризующиеся наличием т.н. свободных валентностей. Активные центры считаются независимыми (т.е. один активный центр не влияет на адсорбционную способность других), и тождественными.
3) Каждый активный центр способен взаимодействовать только с одной молекулой адсорбата; в результате на поверхности может образоваться только один слой адсорбированных молекул.
4) Процесс адсорбции является обратимым и равновесным – адсорбированная молекула удерживается активным центром некоторое время, после чего десорбируется; т.о., через некоторое время между процессами адсорбции и десорбции устанавливается динамическое равновесие.

Рис. 4.5 Изотерма мономолекулярной адсорбции
В состоянии равновесия скорость адсорбции равна скорости десорбции. Скорость десорбции прямо пропорциональна доле занятых активных центров (х), а скорость адсорбции прямо пропорциональна произведению концентрации адсорбата на долю свободных активных центров (1 – х):
 (IV.7)
(IV.7)
 (IV.8)
(IV.8)
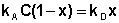 (IV.9)
(IV.9)
Отсюда находим х:
 (IV.10)
(IV.10)
Разделив числитель и знаменатель правой части уравнения (IV.10) на kA, получим:
 (IV.11)
(IV.11)
Максимально возможная величина адсорбции Го достигается при условии, что все активные центры заняты молекулами адсорбата, т.е. х = 1. Отсюда следует, что х = Г / Го. Подставив это в уравнение (IV.11), получаем:
 (IV.12)
(IV.12)
 (IV.13)
(IV.13)
Уравнение (IV.13) есть изотерма мономолекулярной адсорбции, связывающая величину адсорбции Г с концентрацией адсорбата С. Здесь b – некоторая постоянная для данной пары адсорбент-адсорбат величина (отношение констант скоростей десорбции и адсорбции), численно равная концентрации адсорбата, при которой занята половина активных центров. График изотермы адсорбции Ленгмюра приведен на рис. 4.5. Константу b можно определить графически, проведя касательную к изотерме адсорбции в точке С = 0.
При описании процесса адсорбции газов в уравнении (IV.13) концентрация может быть заменена пропорциональной величиной парциального давления газа:
 (IV.14)
(IV.14)
Теория мономолекулярной адсорбции Ленгмюра применима для описания некоторых процессов адсорбции газов и растворенных веществ при небольших давлениях (концентрациях) адсорбата.
Теория полимолекулярной адсорбции Поляни
На практике часто (особенно при адсорбции паров) встречаются т.н. S-образные изотермы адсорбции (рис. 4.6), форма которых свидетельствует о возможном, начиная с некоторой величины давления, взаимодействии адсорбированных молекул с адсорбатом.

Рис. 4.6 Изотерма полимолекулярной адсорбции
Для описания таких изотерм адсорбции М. Поляни предложил теорию полимолекулярной адсорбции, основанную на следующих основных положениях:
1. Адсорбция вызвана чисто физическими силами.
2. Поверхность адсорбента однородна, т.е. на ней нет активных центров; адсорбционные силы образуют непрерывное силовое поле вблизи поверхности адсорбента.
3. Адсорбционные силы действуют на расстоянии, большем размера молекулы адсорбата. Иначе говоря, у поверхности адсорбента существует некоторый адсорбционный объём, который при адсорбции заполняется молекулами адсорбата.
4. Притяжение молекулы адсорбата поверхностью адсорбента не зависит от наличия в адсорбционном объеме других молекул, вследствие чего возможна полимолекулярная адсорбция.
5. Адсорбционные силы не зависят от температуры и, следовательно, с изменением температуры адсорбционный объем не меняется.
Уравнение Фрейндлиха
Теоретические представления, развитые Ленгмюром и Поляни, в значительной степени идеализируют и упрощают истинную картину адсорбции. На самом деле поверхность адсорбента неоднородна, между адсорбированными частицами имеет место взаимодействие, активные центры не являются полностью независимыми друг от друга и т.д. Все это усложняет вид уравнения изотермы. Г. Фрейндлих предположил, что число молей адсорбированного газа или растворенного вещества, приходящееся на единицу массы адсорбента (т.н. удельная адсорбция x/m) должна быть пропорциональна равновесному давлению (для газа) или равновесной концентрации (для веществ, адсорбируемых из раствора) адсорбента, возведенной в некоторую степень, которая всегда меньше единицы:
 (IV.15)
(IV.15)
 (IV.16)
(IV.16)
 (а) (а)
|  (б) (б)
|
Рис. 4.7 Изотерма адсорбции Фрейндлиха в обычных (а) и логарифмических (б) координатах
Показатель степени n и коэффициент пропорциональности а в уравнении Фрейндлиха определяются экспериментально. Логарифмируя уравнения (IV.15 - IV.16), получаем:
 (IV.17)
(IV.17)
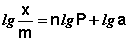 (IV.18)
(IV.18)
Т.о., зависимость логарифма удельной адсорбции от логарифма концентрации (давления) графически выражается прямой линией, отсекающей на оси ординат отрезок, равный lga, тангенс угла наклона которой к оси абсцисс равен по величине показателю степени при давлении или концентрации (рис. 4.7):
 (IV.19)
(IV.19)
4.1.5 Адсорбция на границе твердое тело – раствор
Молекулярная адсорбция из растворов
Изотермы адсорбции растворенных веществ из раствора по своему виду аналогичны изотермам адсорбции для газов; для разбавленных растворов эти изотермы хорошо описываются уравнениями Фрейндлиха или Ленгмюра, если в них подставить равновесную концентрацию растворенного вещества в растворе. Однако адсорбция из растворов является значительно более сложным явлением по сравнению с газовой, поскольку одновременно с адсорбцией растворенного вещества часто происходит и адсорбция растворителя.

Рис. 4.8 Ориентация молекул ПАВ на поверхности адсорбента
Зависимость адсорбции от строения молекул адсорбата очень сложна, и вывести какие-либо закономерности довольно трудно. Молекулы многих органических веществ состоят из полярной (гидрофильной) и неполярной (гидрофобной) группировок, т.е. являются поверхностно-активными веществами. Молекулы ПАВ при адсорбции на твердом адсорбенте ориентируются на его поверхности таким образом, чтобы полярная часть молекулы была обращена к полярной фазе, а неполярная – к неполярной. Так, при адсорбции алифатических карбоновых кислот из водных растворов на неполярном адсорбенте – активированном угле – молекулы ориентируются углеводородными радикалами к адсорбенту; при адсорбции из бензола (неполярный растворитель) на полярном адсорбенте – силикагеле – ориентация молекул кислоты будет обратной (рис. 4.8).
Адсорбция из растворов электролитов
Адсорбция из водных растворов электролитов происходит, как правило, таким образом, что на твердом адсорбента из раствора адсорбируются преимущественно ионы одного вида. Преимущественная адсорбция из раствора или аниона, или катиона определяется природой адсорбента и ионов. Механизм адсорбции ионов из растворов электролитов может быть различным; выделяют обменную и специфическую адсорбцию ионов.
Обменная адсорбция представляет собой процесс обмена ионов между раствором и твердой фазой, при котором твердая фаза поглощает из раствора ионы какого-либо знака (катионы либо анионы) и вместо них выделяет в раствор эквивалентное число других ионов того же знака. Обменная адсорбция всегда специфична, т.е. для данного адсорбента к обмену способны только определенные ионы; обменная адсорбция обычно необратима.
При специфической адсорбции адсорбция на поверхности твердой фазы ионов какого-либо вида не сопровождается выделением в раствор эквивалентного числа других ионов того же знака; твердая фаза при этом приобретает электрический заряд. Это приводит к тому, что вблизи поверхности под действием сил электростатического притяжения группируется эквивалентное число ионов с противоположным зарядом, т.е. образуется двойной электрический слой. Взаимодействие концентрирующихся на поверхности зарядов приводит к понижению поверхностной энергии системы. Для случая специфической адсорбции электролита Песковым и Фаянсом было сформулировано следующее эмпирическое правило (правило Пескова – Фаянса):
На поверхности кристаллического твердого тела из раствора электролита специфически адсорбируется ион, который способен достраивать его кристаллическую решетку или может образовывать с одним из ионов, входящим в состав кристалла, малорастворимое соединение.
4.2 КОЛЛОИДНЫЕ СИСТЕМЫ
Коллоидные системы относятся к дисперсным системам – системам, где одно вещество в виде частиц различной величины распределено в другом (см. разд. 4.1). Дисперсные системы чрезвычайно многообразны; практически всякая реальная система является дисперсной. Дисперсные системы классифицируют прежде всего по размеру частиц дисперсной фазы (или степени дисперсности); кроме того, их разделяют на группы, различающиеся по природе и агрегатному состоянию дисперсной фазы и дисперсионной среды.
Если дисперсионной средой является жидкость, а дисперсной фазой – твердые частицы, система называется взвесью или суспензией; если дисперсная фаза представляет собой капельки жидкости, то систему называют эмульсией. Эмульсии, в свою очередь, подразделяют на два типа: прямые, или "масло в воде" (когда дисперсная фаза – неполярная жидкость, а дисперсионная среда – полярная жидкость) и обратные, или "вода в масле" (когда полярная жидкость диспергирована в неполярной). Среди дисперсных систем выделяют также пены (газ диспергирован в жидкости) и пористые тела (твердая фаза, в которой диспергированы газ либо жидкость). Основные типы дисперсных систем приведены в табл.1.
По степени дисперсности выделяют обычно следующие классы дисперсных систем:
Грубодисперсные системы – системы, размер частиц дисперсной фазы в которых превышает 10-7 м.
Коллоидные системы – системы, размер частиц дисперсной фазы в которых составляет 10-7 – 10-9 м. Коллоидные системы характеризуются гетерогенностью, т.е. наличием поверхностей раздела фаз и очень большим значением удельной поверхности дисперсной фазы. Это обусловливает значительный вклад поверхностной фазы в состояние системы и приводит к появлению у коллоидных систем особых, присущих только им, свойств.
Иногда выделяют молекулярно(ионно)-дисперсные системы, которые, строго говоря, являются истинными растворами, т.е. гомогенными системами, поскольку в них нет поверхностей раздела фаз.
Коллоидные системы, в свою очередь, подразделяются на две группы, резко отличные по характеру взаимодействий между частицами дисперсной фазы и дисперсионной среды – лиофобные коллоидные растворы (золи) и растворы высокомолекулярных соединений (ВМС), которые ранее называли лиофильными коллоидами. К лиофобным коллоидам относятся системы, в которых частицы дисперсной фазы слабо взаимодействуют с дисперсионной средой; эти системы могут быть получены только с затратой энергии и устойчивы лишь в присутствии стабилизаторов.
Растворы ВМС образуются самопроизвольно благодаря сильному взаимодействию частиц дисперсной фазы с дисперсионной средой и способны сохранять устойчивость без стабилизаторов. Лиофобные коллоиды и растворы ВМС различаются также и структурой частиц, составляющих дисперсную фазу. Для лиофобных коллоидов единицей структуры является сложный многокомпонентный агрегат переменного состава – мицелла, для растворов ВМС – макромолекула.
Таблица 1. Основные типы дисперсных систем
| Дисперсная фаза | Дисперсионная среда | Условное обозначение | Примеры дисперсных систем |
| Жидкость | Газ | ж/г | Туман, облака, жидкие аэрозоли |
| Твердое тело | Газ | т/г | Дым, пыль, твердые аэрозоли |
| Газ | Жидкость | г/ж | Пены, газовые эмульсии |
| Жидкость | Жидкость | ж/ж | Эмульсии (молоко, латекс) |
| Твердое тело | Жидкость | т/ж | Суспензии, коллоидные растворы, гели, пасты |
| Газ | Твердое тело | г/т | Твердые пены, пористые тела (пенопласты, силикагель, пемза) |
| Жидкость | Твердое тело | ж/т | Жемчуг, опал |
| Твердое тело | Твердое тело | т/т | Цветные стекла, сплавы |
4.2.1 Методы получения лиофобных коллоидов
Коллоидные системы по степени дисперсности занимают промежуточное положение между истинными растворами (молекулярно- или ионно-дисперсными системами) и грубодисперсными системами. Поэтому коллоидные растворы могут быть получены либо путем ассоциации (конденсации) молекул и ионов истинных растворов, либо дальнейшим раздроблением частиц дисперсной фазы грубодисперсных систем.
Методы получения коллоидных растворов также можно разделить на две группы: методы конденсации и диспергирования (в отдельную группу выделяется метод пептизации, который будет рассмотрен позднее). Еще одним необходимым для получения золей условием, помимо доведения размеров частиц до коллоидных, является наличие в системе стабилизаторов – веществ, препятствующих процессу самопроизвольного укрупнения коллоидных частиц.
Дисперсионные методы
Дисперсионные методы основаны на раздроблении твердых тел до частиц коллоидного размера и образовании таким образом коллоидных растворов. Процесс диспергирования осуществляется различными методами: механическим размалыванием вещества в т.н. коллоидных мельницах, электродуговым распылением металлов, дроблением вещества при помощи ультразвука.
Методы конденсации
Вещество, находящееся в молекулярно-дисперсном состоянии, можно перевести в коллоидное состояние при замене одного растворителя другим – т.н. методом замены растворителя. В качестве примера можно привести получение золя канифоли, которая не растворяется в воде, но хорошо растворима в этаноле. При постепенном добавлении спиртового раствора канифоли к воде происходит резкое понижение растворимости канифоли, в результате чего образуется коллоидный раствор канифоли в воде. Аналогичным образом может быть получен гидрозоль серы.
Коллоидные растворы можно получать также и методом химической конденсации, основанном на проведении химических реакций, сопровождающихся образованием нерастворимых или малорастворимых веществ. Для этой цели используются различные типы реакций – разложения, гидролиза, окислительно-восстановительные и т.д. Так, красный золь золота получают восстановлением натриевой соли золотой кислоты формальдегидом:
NaAuO2 + HCOH + Na2CO3 ––> Au + HCOONa + H2O
Строение мицеллы данного золя можно представить следующей схемой (см. разд. 4.2.2):
{[Au]m· n AuO2–· (n-x) Na+}x– · xNa+
Аналогичным образом получают золь серебра из разбавленных растворов нитрата серебра. Золь серы может быть получен окислением сероводорода кислородом в водном растворе, действием на сероводород сернистого газа либо разложением тиосерной кислоты:
H2S + O2 ––> S + H2O
H2S + SO2 ––> S + H2O
H2S2O3 ––> H2O + SO2 + S
Строение золя серы можно представить схемой:
{[S]m · n HS– · (n-x) H+}x– · x H+
Золи могут быть получены также в результате реакций ионного обмена, в результате которых выделяется нерастворимая соль, образующая при определенных условиях коллоидный раствор; так получают, например, золь иодида серебра (см. ниже).
Процесс гидролиза различных солей может приводить к образованию коллоидных растворов нерастворимых гидроксидов или кислот; так получают, например, золь гидроксида железа(III), имеющий следующее строение:
{[Fe(OH)3]m n FeO+ · (n–x)Cl–}x+ x Cl–
4.2.2 Агрегативная устойчивость лиофобных коллоидов.
Строение коллоидной мицеллы
Лиофобные коллоиды обладают очень высокой поверхностной энергией и являются поэтому термодинамически неустойчивыми; это делает возможным самопроизвольный процесс уменьшения степени дисперсности дисперсной фазы (т.е. объединение частиц в более крупные агрегаты) – коагуляцию золей. Тем не менее золям присуща способность сохранять степень дисперсности – агрегативная устойчивость, которая обусловлена, во-первых, снижением поверхностной энергии системы благодаря наличию на поверхности частиц дисперсной фазы двойного электрического слоя и, во-вторых, наличием кинетических препятствий для коагуляции в виде электростатического отталкивания частиц дисперсной фазы, имеющих одноименный электрический заряд.
Строение структурной единицы лиофобных коллоидов – мицеллы – может быть показано лишь схематически, поскольку мицелла не имеет определенного состава. Рассмотрим строение коллоидной мицеллы на примере гидрозоля иодида серебра, получаемого взаимодействием разбавленных растворов нитрата серебра и иодида калия:
AgNO3 + KI ––> AgI + KNO3
Коллоидная мицелла золя иодида серебра (см. рис. 4.9) образована микрокристаллом иодида серебра, который способен к избирательной адсорбции из окружающей среды катионов Ag+ или иодид-ионов. Если реакция проводится в избытке иодида калия, то кристалл будет адсорбировать иодид-ионы; при избытке нитрата серебра микрокристалл адсорбирует ионы Ag+. В результате этого микрокристалл приобретает отрицательный либо положительный заряд; ионы, сообщающие ему этот заряд, называются потенциалопределяющими, а сам заряженный кристалл – ядром мицеллы. Заряженное ядро притягивает из раствора ионы с противоположным зарядом – противоионы; на поверхности раздела фаз образуется двойной электрический слой. Некоторая часть противоионов адсорбируется на поверхности ядра, образуя т.н. адсорбционный слой противоионов; ядро вместе с адсорбированными на нем противоионами называют коллоидной частицей или гранулой. Остальные противоионы, число которых определяется, исходя из правила электронейтральности мицеллы, составляют диффузный слой противоионов; противоионы адсорбционного и диффузного слоев находятся в состоянии динамического равновесия адсорбции – десорбции.
Схематически мицелла золя иодида серебра, полученного в избытке иодида калия (потенциалопределяющие ионы – анионы I–, противоионы – ионы К+) может быть изображена следующим образом:
{[AgI]m · nI– · (n-x)K+}x– · x K+
При получении золя иодида серебра в избытке нитрата серебра коллоидные частицы будут иметь положительный заряд:
{[AgI]m · nAg+ · (n-x)NO3–}x+ · x NO3–

Рис. 4.9.Строение коллоидной мицеллы
Агрегативная устойчивость золей обусловлена, таким образом, рядом факторов: во-первых, снижением поверхностной энергии дисперсной фазы (т.е. уменьшения движущей силы коагуляции) в результате образования двойного электрического слоя и, во-вторых, наличием кинетических препятствий для коагуляции в виде электростатического отталкивания имеющих одноименный заряд коллоидных частиц и противоионов. Еще одна причина устойчивости коллоидов связана с процессом гидратации (сольватации) ионов. Противоионы диффузного слоя сольватированы; эта оболочка из сольватированных противоионов также препятствует слипанию частиц.
4.2.3 Коагуляция лиофобных коллоидов
Как было показано выше, лиофобные коллоиды являются термодинамически неустойчивыми системами, существующими благодаря стабилизации за счет возникновения двойного электрического слоя. Изменение состояния ДЭС может, следовательно, привести к потере агрегативной устойчивости – слипанию частиц в более крупные агрегаты, т.е. коагуляции золя. Коагуляция золей может быть вызвана различными факторами: прибавлением электролитов, нагреванием или замораживанием, механическим воздействием и т.д. Наиболее важным и изученным фактором коагуляции гидрофобных коллоидов является воздействие на них растворов электролитов.
Для коагуляции золей электролитами установлен ряд эмпирических закономерностей.
1. Для начала коагуляции золя необходима некоторая минимальная концентрация электролита, называемая порогом коагуляции γ.
2. Коагулирующим действием обладает тот из ионов электролита, заряд которого противоположен заряду коллоидных частиц, причем коагулирующее действие иона тем сильнее, чем больше его заряд (правило Шульце – Гарди или правило значности). Величины порогов коагуляции двухзарядных ионов примерно на порядок, а трехзарядных – на два порядка меньше, чем для однозарядных ионов. Правило значности имеет приближенный характер и справедливо только для неорганических ионов; некоторые однозарядные органические ионы обладают более сильным коагулирующим действием, чем двухзарядные неорганические ионы, что обусловлено их сильной специфической адсорбируемостью.
3. В рядах неорганических ионов с одинаковыми зарядами коагулирующее действие возрастает с уменьшением гидратируемости ионов; например, в ряду однозарядных катионов щелочных металлов коагулирующее действие возрастает от лития к рубидию:
γ (Li+) > γ (Na+) > γ (К+) > γ (Rb+)
Ряды, в которые сгруппированы по возрастанию либо по убыванию коагулирующего действия ионы с одинаковым зарядом, называют лиотропными рядами.
4. В осадках, получаемых при коагуляции золей электролитами, всегда присутствуют ионы, вызвавшие коагуляцию.
5. При коагуляции золей смесями электролитов сравнительно редко наблюдается их независимое (аддитивное) действие; обычно имеет место взаимное усиление либо ослабление коагулирующего действия (синергизм либо антагонизм ионов).
Механизм и кинетика коагуляции золей электролитами
Необходимому для коагуляции сближению частиц дисперсной фазы препятствует, как было показано выше, электростатическое отталкивание имеющих одноименный заряд коллоидных частиц и противоионов и взаимодействие сольватных оболочек противоионов диффузного слоя. При добавлении к золю раствора электролита имеющееся равновесие адсорбции – десорбции между противоионами адсорбционного и диффузного слоев смещается в сторону адсорбции вследствие увеличения в дисперсионной среде концентрации ионов, имеющих заряд, противоположный заряду ядра (ионы с одноименным зарядом в равновесии адсорбции – десорбции не участвуют). Адсорбция дополнительного числа противоионов приводит к уменьшению заряда коллоидных частиц, уменьшению числа противоионов диффузного слоя (уменьшению толщины ДЭС) и, следовательно, к снижению агрегативной устойчивости золя. При достижении некоторого предельного значения заряда коллоидные частицы получают возможность сближения и объединения в более крупные агрегаты за счет ван-дер-ваальсовых сил; иными словами, происходит коагуляция золя.
Очевидно, что, поскольку при адсорбции многозарядных противоионов заряд коллоидной частицы уменьшается быстрее, чем при адсорбции того же числа однозарядных противоионов; адсорбируемость неорганических ионов с увеличением их заряда также возрастает. Следствием этого и является тот факт, что величина порога коагуляции для неорганических ионов будет тем меньше, чем больше заряд иона-коагулянта (величина порога коагуляции γ обратно пропорциональна заряду иона-коагулянта в шестой степени z6).
Процесс коагуляции золя характеризуется определенной величиной скорости коагуляции, которую можно определить как изменение числа коллоидных частиц в единице объема за единицу времени. Скорость коагуляции золя электролитами зависит как от концентрации самого золя, так и от концентрации электролитов. Типичный вид коагуляционной кривой (зависимости отношения концентрации коллоидных частиц n к их начальной концентрации nо от времени t) и кривой зависимости скорости коагуляции V от концентрации электролита С показан на рисунках 4.10-4.11. На кривой ОАБВ (рис. 4.11) отрезок ОА отвечает периоду скрытой коагуляции, при которой золь сохраняет свою устойчивость. В точке А при концентрации электролита С1 начинается явная коагуляция; на участке АБ скорость коагуляции быстро возрастает с ростом концентрации электролита. На участке БВ скорость коагуляции остается постоянной; это связано с тем, что при концентрации электролита С2 величина ζ-потенциала становится равной нулю; скорость коагуляции при этом достигает максимального значения.

Рис. 4.10 Коагуляционная кривая. Рис. 4.11 Зависимость скорости коагуляции от концентрации.
Взаимная коагуляция золей
Коагуляция золя может быть вызвана его взаимодействием с другим золем, частицы которого имеют противоположный заряд. Так, смешение золя гидроксида железа, частицы которого имеют положительный заряд, с отрицательно заряженным золем сульфида мышьяка приводит к их взаимной коагуляции:
{[Fe(OH)3]m n FeO+· (n-x)Cl–}x+ xCl–{[Аs2S3]m · n НS–· (n-x)Н+}x– · xН+
В данном случае коагуляция обусловлена тем, что коллоидные частицы одного вида являются как бы очень крупными многозарядными ионами – коагулянтами для частиц другого вида. Взаимная коагуляция коллоидных систем может наблюдаться и тогда, когда частицы золей имеют одноименный заряд; в этом случае причиной потери устойчивости одного из золей является сильная специфическая адсорбция иона – стабилизатора данной системы поверхностью коллоидных частиц другой системы.
Старение золей и пептизация
Термодинамическая неустойчивость лиофобных коллоидных систем является причиной старения золей – самопроизвольной коагуляции (автокоагуляции) золей. Автокоагуляция золей происходит значительно медленнее, чем коагуляция электролитами; так, золи золота могут сохраняться без видимых изменений десятилетиями. Одной из основных причин старения золей является медленно совершающийся процесс перекристаллизации вещества ядра.
Пептизацией (дезагрегацией) называется процесс расщепления коагулировавшего золя (коагулята) на первичные частицы – процесс, противоположный коагуляции. Пептизация возможна лишь тогда, когда структура частиц в коагуляте не изменена по сравнению с первоначальной (т.е. когда еще не произошло полного сращивания частиц и они слабо связаны друг с другом). Различают непосредственную и опосредованную пептизацию.
Непосредственная пептизация происходит в результате добавления к коагуляту электролита, содержащего потенциалопределяющий ион; в результате его специфической адсорбции на поверхности частиц дисперсной фазы их заряд вновь увеличивается, толщина двойного электрического слоя возрастает. Это приводит к тому, что силы отталкивания между частицами начинают преобладать над силами притяжения; происходит деагрегация – распад образовавшегося ранее агрегата из слипшихся частиц.
Опосредованная пептизация вызывается добавлением в систему вещества, химическое взаимодействие которого с поверхностью коагулята приводит к высвобождению потенциалопределяющих ионов. Например, коагулировавший золь гидроксида железа(III) может быть пептизирован добавлением в систему либо какой-либо соли железа (непосредственная пептизация), либо соляной кислоты (опосредованная пептизация).
4.2.4 Двойной электрический слой и электрокинетические явления
При рассмотрении строения мицеллы было показано, что на поверхности лиофобных коллоидов образуется двойной электрический слой. Первая теория строения ДЭС была развита Гельмгольцем и Перреном; в их представлении двойной электрический слой подобен плоскому конденсатору, внутренняя обкладка которого находится в твердой фазе, а внешняя – в жидкости параллельно поверхности ядра на расстоянии порядка диаметра иона. Потенциал электрического поля внутри ДЭС φ в этом случае линейно уменьшается с увеличением расстояния от поверхности r (рис. 4.12а).
Позднее Гуи и Чепмен предложили другую модель, согласно которой противоионы, благодаря тепловому движению, образуют вблизи твердой поверхности ядра диффузную ионную атмосферу. Уменьшение электрического потенциала ДЭС φ с увеличением расстояния r в этом случае происходит нелинейно (рис. 4.12б).

Рис. 4.12 Строение ДЭС: а) – по Гельмгольцу и Перрену, б) – по Гуи и Чепмену, в) – по Штерну. Вверху – схема расположения противоионов, внизу – зависимость потенциала от расстояния
Предложенная Штерном модель строения ДЭС объединяет ранние модели, учитывая как адсорбцию противоионов, так и их тепловое движение. Согласно этой модели, являющейся в настоящее время общепринятой, часть противоионов находится на расстояниях порядка диаметра иона от поверхности ядра, образуя т.н. слой Гельмгольца (адсорбционный слой противоионов), а другая часть образует диффузный слой (т.н. слой Гуи). Потенциал диффузной части двойного электрического слоя называют электрокинетическим потенциалом (см. рис.4.12в). Электрокинетический потенциал обычно обозначают греческой буквой ζ (дзета) и называют поэтому дзета-потенциалом. Поскольку ζ-потенциал пропорционален заряду коллоидной частицы, агрегативная устойчивость золя пропорциональна его величине.
Если поместить золь в постоянное электрическое поле, то, как и в растворах электролитов, заряженные частицы будут двигаться к противоположно заряженным электродам: коллоидная частица с адсорбированными на ней противоионами – в одну сторону, противоионы диффузного слоя – в другую. Сила, с которой электрическое поле действует на частицы и, следовательно, скорость движения частиц, очевидно, будет пропорциональна ζ-потенциалу. Движение частиц дисперсной фазы в электрическом поле называется электрофорезом. Явление электрофореза можно наблюдать, поместив в U-образную трубку какой-либо окрашенный золь, поверх которого налит не смешивающийся с золем бесцветный электролит. Если опустить в электролит электроды и наложить разность потенциалов, то граница окрашенного золя в одном из колен трубки будет подниматься, в другом – опускаться (рис. 4.13). Если поместить в U-образную трубку пористую перегородку (например, мелкий кварцевый песок) и заполнить её водой, то при наложении разности потенциалов в одном колене будет наблюдаться подъем уровня жидкости, в другом – его опускание (рис. 4.14). Движение дисперсной среды в электрическом поле относительно неподвижной дисперсной фазы (в рассмотренном случае – относительно поверхности пористых тел) называется электроосмосом. Явления электрофореза и электроосмоса получили общее название электрокинетических явлений.

| 
|
| Рис. 4.13 Схема опыта по электрофорезу | Рис. 4.14Схема опыта по электроосмосу |
Скорость движения частиц дисперсной фазы при электрофорезе, а также скорость движения дисперсной среды при электроосмосе прямо пропорциональны напряженности электрического поля E и диэлектрической проницаемости дисперсионной среды ε и обратно пропорциональны вязкости среды η. Скорость движения частиц дисперсной фазы при электрофорезе U связана с величиной ζ-потенциала уравнением Гельмгольца-Смолуховского (К – постоянная, зависящая от формы частиц дисперсной фазы; для сферических частиц К = 6):
 (IV.20)
(IV.20)
Обратные электрофорезу и электроосмосу электрокинетические явления (т.н. электрокинетические явления второго рода) называются соответственно потенциал седиментации и потенциал протекания. Потенциал седиментации (эффект Дорна) – возникновение разности потенциалов при вынужденном движении дисперсной фазы относительно неподвижной дисперсионной среды (например, под действием силы тяжести). Потенциал протекания (эффект Квинке) есть явление возникновения разности потенциалов при движении дисперсионной среды относительно неподвижной дисперсной фазы (например, при продавливании электролита через пористое тело).
4.2.5 Кинетическая устойчивость золей. Седиментация
Частицы дисперсной фазы одновременно испытывают действие силы земного притяжения и архимедовой силы; в зависимости от соотношения плотностей дисперсионной среды и дисперсной фазы равнодействующая этих сил будет вынуждать частицы к оседанию либо всплытию. Процесс оседания либо всплытия коллоидных частиц в золе называется седиментацией. Однако седиментации всегда противодействует другой процесс, стремящийся к равномерному распределению коллоидных частиц по всему объему раствора – диффузия, осуществляемая под действием броуновского движения частиц. Соотношение между этими двумя процессами определяет кинетическую устойчивость золей – способность коллоидных частиц удерживаться во взвешенном состоянии, не подвергаясь седиментации.
В статистической теории броуновского движения, развитой А. Эйнштейном, вводится понятие средний сдвиг ±Δx, представляющий собой проекцию расстояния между положениями частицы X1 и X2, в которых частица находилась во время двух последовательных наблюдений через время t. Значение квадрата среднего сдвига можно найти по уравнению Эйнштейна, связывающего Δx2 с температурой T, радиусом взвешенных частиц r и вязкостью среды η:
 (IV.21)
(IV.21)
Средний сдвиг частицы связан с коэффициентом диффузии D, который может быть рассчитан по уравнению (IV.22):
 (IV.22)
(IV.22)
 (IV.23)
(IV.23)
Как видно из уравнения (IV.23), величина коэффициента диффузии определяется отношением тепловой энергии молекул kT и вязкостного сопротивления диффузии со стороны среды. Поскольку процесс диффузии проявляется тем сильнее, чем меньше масса коллоидных частиц, более крупные частицы оседают либо всплывают в первую очередь. Кинетическая устойчивость золя, таким образом, прямо пропорциональна степени дисперсности золя. Заметное оседание частиц в системе, обладающей высокой кинетической устойчивостью, можно вызвать при помощи центрифугирования золя, используя значительные по величине центробежные силы, что многократно увеличивает силу, действующую на частицу и способствующую её оседанию (современные ультрацентрифуги работают при ускорениях свыше 400000g).

Рис. 4.15 Кривая седиментации Рис. 4.16 Кривая распределения
Методы седиментации и ультрацентрифугирования применяются для изучения полидисперсности коллоидных систем, обусловленной существованием в коллоидных системах частиц различных размеров. Изучение полидисперсности коллоидных систем для установления количественного распределения частиц по размерам (т.н. кривых распределения) – седиментационный анализ – производится при помощи измерения возрастания веса осевших частиц w со временем. По результатам такого исследования строят кривые седиментации (рис. 4.15). Проводя анализ кривой седиментации, можно рассчитать кривую распределения для данной системы, которая характеризует относительное содержание в системе частиц разного размера (рис. 4.16). Обычно кривые распределения содержат один максимум, который соответствует rв – наиболее вероятному радиусу частиц дисперсной фазы
4.2.6 Очистка коллоидных систем
Некоторые молекулярно-кинетические свойства коллоидных систем используют для очистки золей от электролитов и молекулярных примесей, которыми полученные золи часто бывают загрязнены. Наиболее распространенными методами очистки коллоидных систем являются диализ, электродиализ и ультрафильтрация, основанные на свойстве некоторых материалов – т.н. полупроницаемых мембран (коллодия, пергамента, целлофана и т.п.) – пропускать ионы и молекулы небольших размеров и задерживать коллоидные частицы. Все полупроницаемые мембраны представляют собой пористые тела, и непроницаемость их для коллоидных частиц обусловлена тем, что коэффициент диффузии для коллоидных частиц значительно (на несколько порядков) меньше, чем для ионов и молекул, имеющих намного меньшие массу и размеры.Прибор для очистки золей методом диализа называется диализатором; простейший диализатор представляет собой сосуд, нижнее отверстие которого затянуто полупроницаемой мембраной (рис. 4.17). Золь наливают в сосуд и помещают последний в ёмкость с дистиллированной водой (обычно проточной); ионы и молекулы примесей диффундируют через мембрану в растворитель.

| 
|
| Рис. 4.17 Схема диализатора | Рис. 4.18 Схема электродиализатора |
Диализ является очень медленным процессом; для более быстрой и полной очистки золей применяют электродиализ. Электродиализатор состоит из трех частей; в среднюю часть, отделенную от двух других полупроницаемыми мембранами, за которыми помещены электроды, наливается золь (рис. 4.18). При подключении к электродам разности потенциалов катионы содержащихся в золе электролитов диффундируют через мембрану к катоду, анионы – к аноду. Преимущество электродиализа заключается в возможности удаления даже следов электролитов (необходимо помнить, что степень очистки ограничивается устойчивостью коллоидных частиц; удаление из золя ионов-стабилизаторов приведет к коагуляции).
Еще одним методом очистки золей является ультрафильтрация – отделение дисперсной фазы от дисперсионной среды путем фильтрования под давлением через полупроницаемые мембраны. При ультрафильтрации коллоидные частицы остаются на фильтре (мембране).
4.2.7 Оптические свойства коллоидных систем
Особые оптические свойства коллоидных растворов обусловлены их главными особенностями: дисперсностью и гетерогенностью. На оптические свойства дисперсных систем в значительной степени влияют размер и форма частиц. Прохождение света через коллоидный раствор сопровождается такими явлениями, как поглощение, отражение, преломление и рассеяние света. Преобладание какого-либо из этих явлений определяется соотношением между размером частиц дисперсной фазы и длиной волны падающего света. В грубодисперсных системах в основном наблюдается отражение света от поверхности частиц. В коллоидных растворах размеры частиц сравнимы с длиной волны видимого света, что предопределяет рассеяние света за счёт дифракции световых волн.
Светорассеяние в коллоидных растворах проявляется в виде опалесценции – матового свечения (обычно голубоватых оттенков), которое хорошо заметно на тёмном фоне при боковом освещении золя. Причиной опалесценции является рассеяние света на коллоидных частицах за счёт дифракции. С опалесценцией связано характерное для коллоидных систем явление – эффект Тиндаля: при пропускании пучка света через коллоидный раствор с направлений, перпендикулярных лучу, наблюдается образование в растворе светящегося конуса.
Процесс дифракционного светорассеяния на частицах, размер которых значительно меньше длины волны описывается уравнением Рэлея, связывающим интенсивность рассеянного единицей объёма света I с числом частиц в единице объёма ν, объёмом частицы V, длиной волны λ и амплитудой А падающего излучения и показателями преломления дисперсной фазы и дисперсионной среды n1 и n2 соответственно:
 (IV.24)
(IV.24)
Из уравнения (IV.18) видно, что, чем меньше длина волны падающего излучения, тем больше будет рассеяние. Следовательно, если на частицу падает белый свет, наибольшее рассеивание рассеяние будут испытывать синие и фиолетовые компоненты. Поэтому в проходящем свете коллоидный раствор будет окрашен в красноватый цвет, а в боковом, отраженном – в голубой.
На сравнении интенсивности светорассеяния золей, один из которых имеет известную концентрацию (степень дисперсности), основан метод определения концентрации либо степени дисперсности золя, называемый нефелометрией. На использовании эффекта Тиндаля основывается ультрамикроскоп – прибор, позволяющий наблюдать коллоидные частицы размером более 3 нанометров в рассеянном свете (в обычном микроскопе можно наблюдать частицы с радиусом не менее 200 нм из-за ограничений, связанных с разрешающей способностью оптики).
Хозяйствующие субъекты без образования юридического лица
Паевой инвестиционный фонд – обособленный имущественный комплекс, состоящий из имущества, переданного в доверительное управление управляю
Дата добавления: 2014-12-03; просмотров: 420; Мы поможем в написании вашей работы!; Нарушение авторских прав |