КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Статические и динамические показатели внешнего дыхания
| Статические показатели внешнего дыхания | Объемные значения показателей внешнего дыхания (литры) |
| Общий объем легких | 5,97 |
| Жизненный объем | 4,78 |
| Дыхательный объем | 0,5 |
| Резервный объем вдоха | 3,28 |
| Резервный объем выдоха | 0,98 |
| Остаточный объем | 1,19 |
| Динамические показатели внешнего дыхания | Значение динамических показателей |
| Частота дыхания в покое | 16 движений/ мин. |
| Максимальный скорость выдоха | 500 л/мин. |
| Максимальная скорость вдоха | 300 л/мин. |
| Максимальный минутный объем | 130 л/мин. |
| Минутный объем альвеолярной вентиляции в покое | 4.9 л/мин. |
| Минутный объем вентиляции мертвого анатомического пространства | 2.1 л/мин. |
Активный выдох (произвольный или непроизвольный при гипоксемии) осуществляется за счет сокращения мышц выдоха (внутренние межреберные и мышцы брюшного пресса). Сокращение внутренних межреберных мышц развивают силы противоположные силам наружных межреберных мышц, что приводит к сокращению объема грудной клетки. Сокращение мышц брюшного пресса повышают внутрибрюшное давление, которое смещает внутренние органы и диафрагму внутрь грудной клетки. Оба действия увеличивают внутригрудное давление, создавая положительный градиент давления и форсированный выход альвеолярного воздуха в атмосферу. После выдоха следует дыхательная пауза (апноэ), которая завершает цикл внешнего дыхания.
Легочная вентиляция зависит от двух параметров: амплитуды (глубины) и частоты дыхания. Вентиляционные параметры легких зависят от анатомических особенностей дыхательного аппарата и количественно оцениваются статическими и динамическими показателями.
Статические и динамические показатели дыхания у молодых здоровых мужчин (средние значение) представлены в таблице 34.2.
34.1.1. Общая этиология и патогенез нарушений легочной вентиляции
Общими причинами нарушения легочной вентиляции являются типические патологические процессы, локализованные как в легких (легочные патологические процессы), так и вне легких. К внелегочным патологическмх процессам относятся те процессы, которые поражают составные элементы аппарата дыхания и, также, итегральные патологические процессы.
34.1.1.1. Итегральные патологические процессы и изменения состава крови
К биохимическим параметрам крови, которые поддерживаются внешним дыханием, относятся следующие: парциальное давление кислорода в артериальной крови (РаО2), парциальное давление углекислого газа в артериальной крови (РаСО2) и концентрация ионов водорода (рН). В свою очередь, эти биохимические параметры крови посредством кибернетической обратной регуляции (feed-back) изменяют внешнее дыхание с целью поддержания гомеостаза внутренней среды.
На уровне моря парциальное давление кислорода в атмосферном воздухе составляет 155 мм рт.ст., в альвеолярном воздухе и артериальной крови – примерно 100 мм рт.ст., а в венозной крови – лишь 40 мм рт.ст. Поддержание в альвеолярном воздухе и в крови более низкого, по сравнению с атмосферой, давления кислорода - это способ защиты клеток организма от токсического действия кислорода.
Содержание углекислого газа в атмосферном воздухе составляет 0,03%, а парциальное давление его – 0,22 мм рт. ст. В тоже время, давление углекислого газа в альвеолярном воздухе и в артериальной крови равно 40 мм рт. ст., а в венозной крови – 46 мм рт. ст. Таким образом, давление углекислого газа в альвеолярном воздухе превышает примерно в 200 раз его давление в атмосфере. Увеличенная концентрация углекислого газа в крови обеспечивает поддержание кислотно-щелочного баланса и значения рН внутренней среды равное примерно 7,36, что является более жизненно важным параметром, чем концентрация кислорода. Можно считать, что легочная вентиляция активно поддерживает постоянно повышенную концентрацию углекислого газа в альвеолах и, соответственно, в крови. Изменения концентрации углекислого газа в альвеолярном воздухе (и в артериальной крови) характеризуют состояние легочной вентиляции: 40 мм рт. ст. – нормовентиляция, > 41 мм рт. ст.- гиповентиляция, < 39 мм рт. ст. - гипервентиляция.
Концентрация ионов водорода в крови выражается отрицательным десятичным логарифмом – рН, который в норме в артериальной крови равен примерно 7,36 (в клетках – 6,9). Исключительно важным является значение внешнего дыхания в быстрой регуляции кислотно-щелочного равновесия посредством интенсификации вентиляции и выделения излишек углекислого газа при ацидотических состояниях или посредством замедления вентиляции и удержания в организме углекислого газа при алкалозных состояниях. В свою очередь, первичные расстройства легочной вентиляции ведут к дыхательному ацидозу или алкалозу.
Биохимические параметры крови, контролируемые внешним дыханием – РаО2, РаСО2, рН, воспринимаются хеморецепторами стенок сосудов, сосредоточенными преимущественно в каротидном клубочке и в аорте. Каротидные и аортальные хеморецепторы в ответ на снижение парциального давления кислорода и рН и на увеличение парциального давления углекислого газа генерируют нервные импульсы, которые передаются через афферентные пути (волокна блуждающего нерва) к дыхательному центру. Каротидный клубочек в 7 раз более чувствителен, чем аортальные рецепторы и его возбуждение одновременно увеличивает частоту и глубину дыхания, в то время как возбуждение рецепторов аортальноой зоны - лишь ускоряет внешнего дыхания. Наряду с периферическими хеморецепторами существует также и рецепторы в мозге – центральные хеморецепторы. Предназначение центральных и периферических рецепторов различно. Так, например, через периферические рецепторы преимущественно реализуется влияние гипоксемии на внешнее дыхание, в то время как гиперкапния и ацидоз действуют преимущественно на центральные рецепторы, которые воспринимают химический состав межклеточной жидкости ствола мозга. В этом контексте, роль периферических рецепторов состоит в поддержании дыхательных рефлексов в условиях острой глубокой гипоксии, когда нервные центры заторможены из-за нехватки энергии и, потому, не реагируют на прямое действие химических раздражителей. Таким образом, периферические рецепторы могут считаться последней структурой дыхательного рефлекса, которая продолжает функционировать при тяжелой гипоксии. Тот факт, что периферические хеморецепторы не реагируют на незначительные изменения рО2 в крови свидетельствует о том, что эти структуры не предназначены для регуляции внешнего дыхания в состоянии покоя или при физической нагрузке, но только для поддержания дыхания в условиях глубокой гипоксии или при нарушениях центральных механизмов дыхания.
Интегральные атологические роцессы существенно влияют на внешнее дыхание. К ним относятся тяжелые расстройства нервной деятельности (мозговая кома), эндокринопатии (гипотироидизм, гипокортицизм, гипер – и гипоинсулинизм), почечная недостаточность, печеночная недостаточность, недостаточность кровообращения, анемии, нарушения метаболизма (гипогликемия, гиперкетонемия), нарушения водного баланса (эксикоз, отек мозга), электролитные нарушения (гипонатриемия, гиперкалиемия), изменения осмотического давления (гиперосмия, гипоонкия), нарушения кислотно-щелочного равновесия (ацидоз, алкалоз), дистермии (гипо – и гипертермия). Экзогенной причиной нарушения легочной вентиляции являются изменения состава атмосферы – гипоксия и гиперкапния атмосферы.
Общим патогенетическим знаменателем перечисленных выше патологических процессов является гипоксемия, гиперкапния, гипер-Н-иония, а возможный финальный эффект – паралич дыхательного центра, остановка внешнего дыхания (апноэ).
Гипоксемия представляет собой уменьшение напряжения кислорода в артериальной крови ниже 50 мм рт.ст. Гипоксемия увеличивает легочную вентиляцию, хотя в меньшей степени, чем чистая гиперкапния или гиперкапния в комбинации с гипоксией. Глубокая гипоксемия приводит к угнетению дыхательного центра и к остановке дыхания – апноэ. В связи с более высокой, по сравнению с кислородом, чувствительностью дыхательного центра к углекислому газу, чрезмерное выведение его из организма при гипервентиляции с установлением гипокапнии уменьшает возбудимость дыхательного центра, что подавляет легочную вентиляцию и вызывает даже апноэ. Это происходит при гипоксии, ассоциированной с гипокапнией, при гипероксемии (повышение давления кислорода в крови), при вдыхании чистого кислорода. Это вызывает гипероксию с одновременной гипокапнией. Сочетание гипероксии с гипокапнией еще больше снижает реактивность дыхательного центра и может привести даже к его торможению. В таких случаях для поддержания возбудимости дыхательного центра рекомендуется ингаляция карбогена – смеси газов, состоящей из 94% кислорода и 6% углекислого газа.
Гиперкапния представляет собой увеличение напряжения углекислого газа в артериальной крови (выше 46 мм рт. ст.). Гиперкапния является результатом его возросшей продукции либо затруднения его выведения из организма. Гиперкапния является самым сильным возбудителем дыхательного центра, вызывая гипервентиляцию, в то время как гипокапния вызывает гиповентиляцию, вплоть до остановки дыхания. Например, повышение давления углекислого газа в артериальной крови с 40 до 60 мм рт. ст. увеличивает объем легочной вентиляции с 7 л/мин до 65 л/мин, а давление углекислого газа в крови равное 70 мм рт. ст. является максимально переносимым и увеличивает легочную вентиляцию до 75 л/мин. Концентрация СО2 свыше 70 мм рт. ст. вызывает паралич дыхательного центра и остановку дыхания. Снижение давления углекислого газа в крови уменьшает реактивность дыхательного центра также и для других раздражителей (включая гипоксию) вплоть до паралича дыхательного центра и остановки легочной вентиляции.
Н+-гипериония (ацидоз) представляет собой повышение концентрации ионов водорода в крови. Постоянство концентрации ионов водорода в крови поддерживается различными гомеостатическими механизмами, одним из которых является легочная вентиляция, что обеспечивает выделение избытка углекислого газа. Дыхательный центр исклютельно чувствителен к колебаниям рН – уменьшение этого параметра на 0,1 единицы возбуждает дыхательный центр и увеличивает легочную вентиляцию на 2 л/мин., в то время как повышение значения рН приводит к подавлению дыхательного центра и гиповентиляции. Следует отметить, что параллельно с непосредственным действием, ионы водорода влияют на дыхательный центр и посредством выделения углекислого газа из бикарбонатов плазмы крови, что также вызывает гиперкапнию и гипервентиляцию.
Гипоксемия, гиперкапния, ацидоз различного происхождения ведут к реактивным изменениям внешнего дыхания: одышка, глубокое и ускоренное дыхание, периодическое дыхание, апноэ, легочная.
Изменения внешнего дыхания, как ответ на изменение биохимического состава крови, первоначально имеют адекватный гомеостатический характер и направлены на поддержание гомеостаза посредством приведения внешнего дыхания в соответствии с потребностями организма. Следует подчеркнуть, что даже адаптивные или компенсаторные легочные реакции могут привести к различным дисгомеостазам – дыхательному алкалозу, одновременно с повышением проницаемости кровеносных сосудов мозга, повышением внутричерепного давления и отеку мозга. Предельные изменения состава крови ведут к апноэ – клинической смерти.
34.1.1.2. Патологические процессы в рефлекторной дуге
внешнего дыхания
Периферические рецепторы являются источником афферентного возбуждения и поддерживают активность дыхательного центра. Дефицит афферентных импульсов обнаруживается у недоношенных новорожденных детей и проявляется асфиксией. В таких случаях необходима дополнительная афферентация, например, посредством механического раздражения кожи ягодиц, ножек чтобы инициировать первый вдох. Напротив, избыток афферентных импульсов вызывает частое, но поверхностное дыхание с увеличением доли вентиляции мертвого анатомического пространства и соответствующим уменьшением альвеолярной вентиляции. В качестве источника чрезмерной афферентации могут служить патологические процессы, локализованные в брюшине, легких, коже.
Дыхательный центр, характеризуется пейсмекерной деятельностью – способностью спонтанно генерировать эфферентные нервные импульсы, под влиянием которых происходит инициация внешнего дыхания – вдоха и выдоха. Частота импульсов, генерированных дыхательным центром, модулируется периферическими нейрорецепторами - хеморецепторами, которые воспринимают биохимические параметры крови (давление О2, СО2, концентрацию ионов водорода) и механорецепторами дыхательных мышц, воздухоносных путей, плевры. Таким образом, активность дыхательного центра приводится в соответствии с актуальными потребностями организма и сохраняется гомеостаз биохимических параметров.
Нарушение деятельности дыхательного центра может произойти из-за патологических процессов, повреждающих любую часть дыхательного рефлекса: нейрорецепторы, афферентные пути, нервные центры, эфферентные пути. Непосредственными причинами нарушения активности дыхательного центра являются прямые его повреждения (энцефалиты, повышенное внутричерепное давление, черепно-мозговые травмы, тяжелые гипоксии, шок, кома, передозировка снотворных и седативных средств, наркоз, наркотики).
К нарушениям дыхательного центра относятся снижение его возбудимости, паралич.
Нарушения активности дыхательного центра проявляются в виде первичной гиповентиляции, ночного апноэ (остановка дыхания во сне), апнейзиса, периодического дыхания, остановки дыхания. Нужно отметить, что первичные повреждения дыхательного центра приводят к нарушению легочной вентиляции при сохранении всего функционального потенциала дыхательного аппарата (дыхательных мышц, грудной клетки, плевры, воздухоносных путей и легочной паренхимы), однако этот потенциал невостребован.
Нервно – мышечный дыхательный аппарат (дыхательная “помпа”, жизненная “помпа”) включает межреберные нервы и мышцы, диафрагмальный нерв и диафрагму и может быть поврежден на уровне центральной и периферической нервной системы, на уровне нервно – мышечных соединений или непосредственно на уровне дыхательных мышц.
Паралич диафрагмы. Диафрагма является самой важной дыхательной мышцей и имеет наибольшее (после сердца) жизненное значение для человека. Диафрагма иннервирована диафрагмальным нервом, берущим начало от С4 (частично от С3 и очень редко от С5).
Дисфункции диафрагмы являются следствием нейрогенных нарушений (прерывание передачи импульсов из нервной системы вследствие травм спинного мозга, сирингомиелии, полиомиелита) и нарушений проводимости диафрагмального нерва (травма, хирургическое вмешательство на грудной клетке и сердце, радиотерапия, опухоли - 30% случаев, нейроинфекции, аневризма аорты, выпотной плеврит, загрудинный зоб, герпес, уремия, инфекци, сахарный диабет). Нарушения функции диафрагмы могут быть вызваны также различными врожденными анатомическими дефектами (диафрагмальная грыжа со смещением брюшных органов в грудную полость). Все перечисленные повреждения могут вызвать односторонний либо двухсторонний паралич диафрагмы.
Патологические процессы, повреждающие нервно-мышечные соединения диафрагмы и межреберных мышц – это отравление антихолинэстеразными препаратами и продуктами бытовой химии (инсектициды), курареподобными веществами, ботулинистическим токсином, а также невриты и миозиты.
Любые нарушения функции диафрагмы и межреберных мышц вызывают изменения вентиляции посредством ограничения экскурсий грудной клетки и неспособности создавать отрицательное внутригрудное давление, достаточное для осуществления вдоха. Функциональная неспособность межреберных мышц компенсируется диафрагмой, в то время как отсутствие сокращений диафрагмы не могут быть компенсированы межреберными мышцами. Это обусловлено тем, что при повреждении диафрагмы во время вдоха, вызванного сокращением межреберных мышц, происходит смещение парализованной диафрагмы и брюшных органов в полость грудной клетки, что делает невозможным процесс вдоха. Организм человека не обладает другими механизмами, способными компенсировать дыхание, нарушенное обездвиженной диафрагмой, поэтому двухсторонний паралич диафрагмы ведет к серьезным нарушениям дыхания – рестриктивная недостаточность вентиляции со снижением общей и жизненной емкости легких до 50 % и к асфиксии. Односторонний паралич диафрагмы часто протекает бессимптомно.
34.1.1.3. Патологические процессы в грудной клетке.
Внепаринхимальная рестрикция легких
Примечательным свойством дыхательного аппарата является податливость (растяжимость, способность к расширению), которая позволяет расширение грудной клетки и прием атмосферного воздуха при вдохе. Общая растяжимость дыхательного аппарата является алгебраической суммой растяжимости грудной клетки и легких. Она зависит от любых изменений в грудной клетке, плевре и легких. Так как объем вдыхаемого воздуха прямо зависит от степени растяжимости дыхательного аппарата, её уменьшение ведет к рестриктивной дыхательной недостаточности.
Легочная рестрикция возможна при уменьшении общей растяжимости дыхательного аппарата за счет преимущественного уменьшения растяжимости грудной клетки (экстрапаренхимальная легочная рестрикция), либо за счет преимущественного уменьшения растяжимости легких (внутрипаренхимальная легочная рестрикция). Легочная рестрикция любого происхождения сопровождается сокращением экспансии легких и уменьшением статических и динамических дыхательных показателей.
Экстрапаренхимальная легочная рестрикциявызвана повреждениями грудной клетки, нервно-мышечного аппарата, плевры. При рестриктивных нарушениях наблюдается уменьшение полной растяжимости дыхательного аппарата, что ведет к уменьшению легочных объемов.
Повреждения грудной клетки, которые чаще всего приводят к нарушению вентиляции - это кифосколиоз, анкилозный спондилит, ожирение, торакопластика.
Повреждения плевры.Плевра (висцеральный и париетальный листки) образует герметически закрытую полость, которая, посредством изменений внутриплеврального давления, осуществляет экскурсию легких. Повреждения плевры нарушают герметичность плевральной полости либо вызывают внутриплевральную гипертензию. В обоих случаях имеет место сдавление или даже спадение легкого, рестрикция их экскурсий с нарушением вентиляции. К наиболее частым повреждениям плевры относятся плевральный выпот, пневмоторакс, гемоторакс, опухоли.
Плевральный выпот. В норме, плевральная полость содержит примерно 1 мл жидкости, которая формируется из равновесия между фильтрационными силами (гидростатическое давление в кровеносных сосудах висцеральной и париетальной плевры) и силой резорбции (онкотическое давление в сосудах крови и давление интерстициальной жидкости, зависящее от лимфатического дренажа). Плевральный выпот представляет собой дисбаланс этих сил с преобладанием фильтрации плазмы над резорбцией фильтрата и лимфатического дренажа. Выпот представлен транссудатом и экссудатом.
Транссудатпредставляет собой скопление ультрафильтрата плазмы крови в плевральной полости вследствие врожденных пороков сердца, цирроза печени, ателектатической болезни, нефротического синдрома, перитонеального диализа, микседемы, облитерирующего перикардита. Транссудат имеет характерные физико-химические свойства - прозрачный или опалесцирующий, низкая вязкость, содержит до 3% белков, небольшое количество клеток, стерилен.
Экссудат образуется при воспалительных процессах (плевриты любой этиологии, парапневмонии), злокачественных опухолях, эмболии легочной артерии, сосудистых коллагенозах, туберкулезе, саркоидозе, асбестозе, панкреатите, травме, перфорации пищевода, радиационном плеврите. Дифференциация экссудата от транссудата основывается на определении физико-химических, биохимических и биологических свойств и имеет диагностическое значение. Так, экссудат характеризуется абсолютной концентрацией белков более 3%, содержанием серопротеинов более 50% от концентрации белков в плазме крови, активностью лактатдегидрогеназы более 60% от её активности в плазме крови, содержанием холестерина более 45 мг/дл. Экссудат характеризуется высоким содержанием клеток (лейкоцитов) и, как правило, инфицирован патогенными микроорганизмами, вызвавшими воспаление. В случае установленного экссудата, необходим дифференцированный цитологический анализ, окраска по Грамму, бактериологический анализ для получения дополнительной информации об этиологии воспалительного процесса.
Пневмоторакс представляет собой наличие в плевральной полости воздуха, проникшего через повреждения грудной клетки или через поврежденный бронх, сообщающийся с плевральной полостью. Сообщение плевральной полсти с атмосферой выравнивает давление воздуха в альвеолах с давлением в атмосфере, затрудняя или делая невозможным вдох (при двустороннем пневмотораксе).
Наличие жидкости (транссудата, экссудата, крови) или воздуха в плевральной полости уменьшает экскурсию легких, уменьшая статические и динамические показатели внешнего дыхания (дыхательный объем, резервный объем вдоха, минутный объем дыхания) и, в дальнейшем, приводит к дыхательной недостаточности.
Подытоживая изложенное, необходимо отметить следующее. При первичных поражениях нервно-мышечного аппарата, грудной клетки и плевры имеет место уменьшение эффективности дыхательных движений, уменьшение растяжимости структур дыхательного аппарата, и, в конце концов, происходит уменьшение легочной вентиляции. В этих случаях первоначально функция дыхательного центра сохранена. Впоследствии, когда изменяется газовый состав крови и кислотно-щелочной баланс, нарушается и функция дыхательного центра, что усугубляет нарушения вентиляции. При первичных рестриктивных нарушениях проходимость дыхательных путей, альвеолярно-капиллярная диффузия, а также перфузия легких не нарушены. Впоследствии, однако, нарушаются и эти функции - происходит рефлекторный спазм сосудов в слабо вентилируемых альвеолах, что ведет к гипоперфузии этих альвеол, изменяется структура альвеолярной стенки с нарушением диффузии, присоединяется воспалительный процесс, который приводит к обструкции дыхательных путей. Таким образом, в финале устанавливаются комплексные процессы со смешанными рестриктивными, обструктивными, диффузионными и перфузионными нарушениями.
34.1.1.4 Патологические процессы в легочной паренхиме. Легочная интрапаренхимальная рестрикция
Пространством эффективной легочной вентиляции являются альвеолы – диффузионная единица дыхательного аппарата. Общее количество альвеол увеличивается от 10 млн при рождении ребенка до, приблизительно, 300 млн у взрослых. Одновременно, с возрастом происходит и увеличение в объеме существующих альвеол. Совокупность легочных альвеол со средним диаметром 0,25 мм и капилляров малого круга образует контакт крови с воздухом с общей площадью ок. 80 м2.
Альвеолы, как и все структуры грудной клетки, обладают двумя основными качествами: растяжимостью и эластичностью.
Растяжимость – это способность растягиваться под действием приложенной центробежной силы, что позволяет увеличение объема и наполнение их атмосферным воздухом при вдохе. Уменьшение растяжимости ведет к сокращению экскурсии легких – наступает рестрикция легких с рестриктивными нарушениями вентиляции.
Второй характерной особенностью альвеол является эластичность – способность возвращаться к первоначальной форме и объему после того, как они были подвержены деформации во время вдоха. Эластичность альвеол обусловлена их собственной эластичностью (благодаря наличию в их стенках эластических волокон) и поверхностным натяжением жидкости, покрывающей их сурфактанта. Сурфактант – это вещество фосфолипидной природы, которое уменьшает поверхностное натяжение на границе альвеола-воздух. Благодаря сурфактанту, эластичность альвеол становится величиной непостоянной, изменяющейся при вдохе и выдохе. Так, при наполнении воздухом и растяжении альвеол, их поверхность увеличивается, а концентрация сурфактанта уменьшается (при этом неизменное количество сурфактанта распределено на большей альвеолярной поверхности). Это увеличивает поверхностное натяжение и эластическую центростремительную силу, что препятствует чрезмерному расширению альвеол.
При выдохе процессы идут в обратном направлении: выход воздуха из альвеол ведет к уменьшению их объема и площади поверхности, а концентрация сурфактанта растет, так как общее неизменное количество сурфактанта распределяется на меньшей альвеолярной поверхности. Это уменьшает поверхностное натяжение и эластическую силу альвеол, что препятствует их спадению и слипанию. Благодаря этому механизму, даже при максимальном выдохе, стенки альвеол не прилипают друг к другу, а в альвеолах сохраняется определенный объем воздуха, называемый остаточным.
Внутрипаренхимальная рестрикция легких представляет собой уменьшение общей растяжимости дыхательного аппарата за счет сокращения растяжимости и эластичности легких. Встречается при диффузных поражениях легких, в результате которых устанавливается избыточная эластическая сила легких, что и ведет к уменьшению всех легочных объемов.
Итак, рестриктивное уменьшение легочной вентиляции является результатом обратимого или стойкого уменьшения эластичности и растяжимости легочной паренхимы.Процессы, вызывающие рестрикцию легких многочисленны. К ним относятся: системные заболевания (коллагенозы – склеродермия, полимиозит, дерматомиозит, системная красная волчанка, ревматоидный артрит, анкилозирующий спондилит); некоторые медикаменты (нитрофураны, золото, циклофосфамид, метотрексат); первичные заболевания легких (саркоидоз, легочный васкулит, альвеолярный протеиноз, эозинофильная пневмония, облитерирующий бронхиолит, организация пневмонии); инфильтрация легких неорганической пылью (силикоз, асбестоз, пневмокониоз, бериллиоз); фиброз легких, вызванный тяжелыми металлами, органической пылью, идиопатический пневмофиброз, после острой интерстициальной пневмонии, интерстациальной лимфоцитарной пневмонии, радиотерапии. Дополнительно к уменьшению общего и дыхательного объема легких, фиброзирующие процессы создают дисбаланс межу вентиляцией и перфузией, нарушают диффузию кислорода, создают внутрилегочное шунтирование, что поддерживают умеренную гипоксемию даже в покое и тяжелую гипоксемию при физической нагрузке. В ответ на гипоксемию появляется легочная гипервентиляция, обеспечиваемая, преимущественно, увеличением частоты дыхания, т.к., функциональные объемы легких уменьшены. Более поздними последствиями являются воспаление и прогрессирующее фиброзирование паренхимы легких, уменьшение васкуляризации одновременно с повышением периферического сопротивления в малом круге, легочная гипертензия, легочное сердце.
Рестрикция легких различного генеза ведет к рестриктивной дыхательной недостаточности. Как правило, рестриктивные процессы характеризуются уменьшением функционального остаточного объема легких (FRC, с англ. Functional rezidual capacity). FRC – это объем воздуха, остающегося в легких после спокойного выдоха, когда дыхательные мышцы полностью расслаблены, а движение потока воздуха остановлено. Таким образом, FRC представляет собой сумму резерва выдоха и остаточного объема. Значение FRC определяется равновесием между центростремительной эластической силой легких и центробежной эластической силой грудной клетки. Фиброзирующие поражения легких характеризуются уменьшением FRC и других легочных объемов, являясь исходом поражения легких, плевры или структур грудной клетки.
Рестрикция легких ведет к уменьшению их наполнения воздухом, и, соответственно, уменьшению вентилируемой альвеолярной поверхности, доступной для газообмена. Тяжесть рестриктивных нарушений соответствует степени уменьшения общего объема, жизненной емкости, дыхательного объема и функционального остаточного объема легких, при сохранении проходимости дыхательных путей. В итоге, уменьшается общая диффузионная способность легких и, одновременно, увеличивается сопротивление сосудов малого круга, дыхание становится частым и поверхностным. В случае, когда рестриктивные нарушения вызваны альтерацией легочной паренхимы, паралелльно нарушается и трансальвеолярный газообмен, что клинически проявляется дефицитом кислорода в крови, особенно при физической нагрузке.
Пневмосклероз – типический патологический процесс, характеризуемый избыточным ростом соединительной ткани в интерстиции легких – межальвеолярных перегородках и смежных структурных, включая кровеносные сосуды (см. гл. 14, том 1).
Причинами пневмосклероза могут быть воспалительные процессы в легочной паренхиме (пневмонии), нарушение крово- и лимфообращения (продолжительная венозная гиперемия, стаз крови или лимфостаз), инфаркт легких, пропитывание ксенобионтами – антракоз, силикоз, асбестоз, острый респираторный дистресс – синдром, аллергические воспаления и др.
В патогенезе пневмосклероза (фиброза) участвует много факторов, но наиболее частыми является воспаление легочной паренхимы (пневмониты, альвеолиты). Клетки, участвующие в воспалении (лимфоциты, макрофаги, нейтрофилы) выделяют цитокины, активирующие размножение фибробластов и избыточное образование коллагеновых волокон.
Пневмосклероз нарушает все функции дыхательного аппарата – вентиляцию, диффузию, перфузию. Таким образом, избыточный рост соединительной ткани уменьшает как растяжимость, так и эластичность легочной паренхимы, одновременно вызывая уменьшение дыхательного объема, гиповентиляцию, увеличение остаточного объема. Также снижается диффузионная способность фиброзированного альвеолярно-капиллярного барьера, уменьшается общая площадь диффузии, а, в дальнейшем, при вовлечении в процесс бронхов, наблюдается их обструкция, вызывающая обструктивные нарушения вентиляции. Фиброзирование кровеносных сосудов ведет к уменьшению площади общего поперечного сечения сосудов малого круга с легочной гипертензией, а, в дальнейшем, и к легочному сердцу.
Эмфизема легких. Эмфизема легких – это стойкое избыточное расширение воздушных пространств легких дистальнее терминальных бронхиол. При эмфиземе легких наблюдается деструкция фибриллярного каркаса альвеолярной стенки, их чрезмерное расширение, разрушение и уменьшение общего количества альвеол, уменьшение общей поверхности диффузии, растяжение капилляров малого круга.
В настоящее время, в патогенезе эмфиземы легких принята гипотеза нарушения равновесия содержания в легких протеиназ и антипротеиназ. Первичной причиной нарушения баланса протеиназы/антипротеиназы может быть врожденный либо приобретенный дефицит антипротеазных ферментов или повышение протеазной активности в альвеолах. Согласно этой гипотезе, разрушение легочной паренхимы может быть следствием снижения антипротеазной защиты легких, избытка выделяемых в легких протеиназ либо комбинированным действием обоих факторов. Таким образом, эмфизема представляется результатом нарушения равновесия между протеиназами и антипротеиназами в сторону преобладания протеиназ.
В норме, в крови циркулирует определенное количество ферментов, включая протеазы экзокринных пищеварительных желез (главным образом, поджелудочной железы). Эти циркулирующие ферменты диффундируют из крови и накапливаются в легочной паренхиме. Другим источником ферментов для паренхимы легких являются фагоциты (особенно полиморфноядерные лейкоциты), количество которых значительно возрастает при воспалительных процессах в легких. Таким образом, в легочной паренхиме появляется протеазный потенциал, представленный панкреатическими протеазами из системного кровотока, а также коллагенозой, эластазой и другими протеазами, продуцированными нейтрофилами и мононуклеарными фагоцитами. Эти ферменты разрушают межклеточное вещество паренхимы легких (эластические и коллагеновые волокна, основное вещество), уменьшают эластичность альвеол и вызывают эмфизему.
Протеазный потенциал легочной паренхимы прямо пропорционален интенсивности воспалительного процесса и усиливается провоспалительными факторами (например, сигаретным дымом).
Повреждающему действию протеолитических ферментов в легких противостоит антипротеазная система, представленная различными антиферментами, которые подавляют протеолитическую активность, поддерживая целостность альвеолярной паренхимы. Основной их функцией является инактивация протеаз, продуцируемых нейтрофилами и выделяемых в легочный интерстиций при воспалительных процессах (трипсина, эластазы, протеазы З, катепсина G). Эластаза нейтрофилов представляет собой основную протеиназу, ответственную за деструкцию альвеол.
Общая антипротеазная активность альвеол представлена почти исключительно (около 95%) альфа-1-антитрипсином (ААТ). ААТ синтезируется, в основном, гепатоцитами; после выхода из печени он циркулирует в несвязанном виде в крови до диффузии в интерстициальную и альвеолярную жидкость. Дефицит ААТ может быть наследственным и приобретенным.
Наследственный дефицит ААТ – одно из наиболее распространенных врожденных заболеваний представителей белой расы, которое встречается с частотой 1:3–5 тыс. (надо отметить, что среди летальных генетических дефектов, дефицит ААТ стоит на первом месте. Генетический дефект проявляется неспособностью печени синтезировать ААТ, низким содержанием ААТ в плазме, и, как следствие, сниженным содержанием его в альвеолах. Было установлено, что содержание ААТ в плазме ниже 20–53 ммоль/л предрасполагает в эластолизу с ранней панацинарной эмфиземой, а достоверный риск эмфиземы появляется при уровне ААТ в плазме ниже 1 ммоль/л.
Основной причиной приобретенной недостаточности ААТ является курение. Повреждающее действие сигаретного дыма заключается в инициации воспалительных процессов в легочной паренхиме с эмиграцией лейкоцитов, секретирующих протеолитические ферменты, в прямом угнетении ААТ, в повреждении ресничек эпителия бронхов, гиперплазии и гиперсекреции бронхиальных желез. Сигаретный дым – это единственный экзогенный фактор, с доказанным риском развития эмфиземы легких. У курильщиков риск развития эмфиземы легких в 2,8 раз больше, чем у некурящих. Увеличение смертности от эмфиземы было достоверно установлено у курильщиков со стажем более 20 лет.
Другими экзогенными факторами развития эмфиземы легких являются внутривенные инфузии медикаментов, содержащих волокна целлюлозы, тальк (например, метадон, метилфенидат), кокаин, героин, иммунодефициты различного генеза, хронические инфекции, СПИД, васкулиты, заболевания соединительной ткани.
При отсутствии ААТ в альвеолах нарушается равновесие между протеазами и антипротеазами с последующим нарушением целостности альвеолярной стенки, что снижает механическую прочность и эластическую тягу альвеол. Уменьшение эластичности делает невозможным возврат альвеол на выдохе к первоначальному объему, что вызывает чрезмерное их расширение, увеличение остаточного объема, то-есть воздуха, который не может быть выведен из альвеол даже при максимальном форсированном выдохе. Соответственно, пропорционально увеличению остаточного объема, уменьшается дыхательный объем и жизненная емкость легких – таким образом, устанавливается эмфизема легких.
Эмфизема легких подразделяется на центрацинарную, панацинарную и парасептальную.
Центроацинарная эмфизема легких начинается в дыхательных бронхиолах и распространяется дистально. Эта форма эмфиземы, названная также центролобулярной, связана с курением и развивается преимущественно в верхних отделах легких.
Панацинарная эмфизема легких равномерно разрушает все близлежащие альвеолы и локализуется преимущественно в нижних отделах легких. Наблюдается у гомозиготных пациентов с дефицитом ААТ.
При парасептальной (дистальной ацинарной) эмфиземе легких повреждаются преимущественно дистальные воздухносные пути и альвеолярные мешки. Процесс локализуется вокруг легочных перегородок или плевры. Хотя воздухообмен сохранен, верхушечные эмфизематозные пузыри могут привести к спонтанному пневмотораксу.
Эмфизема легких, как правило, сопровождается хроническим бронхитом, вследствие чего патологические изменения появляются не только в легочной паренхиме, но и в средних и крупных бронхах. Хронический бронхит характеризуется увеличением в объеме и усилением секреции желез слизистой оболочки бронхов, очагами плоскоклеточной метаплазии слизистой бронхов, воспалением и утолщением их стенок, дисфункцией ресничек (гипо- или акинезия ресничек), гиперплазией гладких мышц бронхов. В дыхательных бронхиолах, пораженных одновременно с более крупными бронхами, происходит мононуклеарное воспаление с закупоркой просвета слизистыми пробками, клеточной метаплазией, гиперплазией гладкой мускулатуры, фиброзом и деформацией. Таким образом, эмфизема легких и воспаление мелких дыхательных путей всегда встречаются вместе. Все это, наряду с потерей эластического каркаса альвеол, нарушают вентиляцию легких.
Эмфизема легких характеризуется увеличением остаточного объема легких, уменьшением резервного объема выдоха, форсированным выдохом в покое (экспираторная одышка). При эмфиземе имеет место «клапанный» обтурационный механизм – при вдохе слизистая пробка из бронхиолы аспирируется в альвеолы, не мешая вдоху, а при выдохе возвращается в бронхиолы, затрудняя его. При стойкой хронической эмфиземе легких эти изменения становятся необратимыми, что вызывает морфологические изменения в легких, вплоть до пневмосклероза.
При умеренной эмфиземе уменьшение вентиляции в большей степени связано с потерей эластичности, чем с воспалением. Напротив, при далекуо зашедшем процессе уменьшение вентиляции в большей степени связано с изменением свойств бронхиол.
При эмфиземе легких, одновременно с разрушением альвеол, изменяются также и сосуды малого круга. Так, в интиме артерий и артериол появляются аномальные продольные мышечные волокна с утолщением мышечной оболочки и фиброзированием интимы. Расширение бронхиальных вен может привести к шунтированию вен большого круга с левым предсердием.
Патогенетическая коррекция нарушенного равновесия протеазы/антипротеазы состоит в угнетении воспалительных явлений в альвеолах, генерирующие протеазы, лечение астмы, предупреждение и лечение респираторных инфекций, стимулирование продукции ААТ либо заместительная терапия этим препаратом. В настящее время для лечения дефицита ААТ предлагается заместительная терапия антиэнзимами, к примеру, еженедельные внутривенные инфузии проластина, который содержит человеческие плазматические белки, в том числе и фракции антипротеаз. Следует отметить, что для курильщиков единственной возможностью продления жизни является отказ от этой привычки.
Ателектаз. Ателектаз (гр. аteles и eкtasis – полное бездействие) – уменьшение объема, спадение и прекращение вентиляции части или всего легкого. В зависимости от этиологии ателектаз делится на обструктивный и необструктивный.
Обструктивный ателектаз – это результат обструкции долевых или сегментарных бронхов (соответственно возникает долевой или сегментарной ателектаз). Обструкция бронхов приводит к прекращению вентиляции части пораженного легкого, газы из альвеол абсорбируются в кровь, альвеолы спадаются. В начале процесса перфузия спавшегося участка продолжается еще какое-то время, но, в отсутствии вентиляции, региональная гипоксемия приводит к рефлекторной вазоконстрикции сосудов невентилируемых альвеол. Это уменьшает до минимума перфузию ателектазированного участка с перераспределением крови к вентилируемым альвеолам.
Необструктивный ателектаз может быть следствием многих факторов. К ним относятся: а) потеря контакта между париетальной и висцеральной плеврой, наличие в плевральной полости воздуха, экссудата, транссудата, крови (пассивный ателектаз); б)повышение внутриплеврального давления, сдавление легких (компрессионный ателектаз); в)отсутствие сурфактанта, острый респираторный дистресс, лучевая пневмония, травма легкого, пневмосклероз и инфильтративные поражения легких, которые увеличивают поверхностное натяжение альвеол, снижая их растяжимость и провоцируя спадение этих альвеол (адгезивный ателектаз).
Из-за отключения участка легкого от вентиляции уменьшается дыхательный объем и возрастает объем функционального мертвого пространства – так возникает гиповентиляция с гипоксемией и гиперкапнией. В невентилируемых участках кровеносные сосуды спазмируются (рефлекторная вазоконстрикция), что приводит к нарушению равновесия вентиляции-перфузии, что усугубляет гипоксемию. Кроме того, вазоконстрикция в малом круге кровообращения приводит к легочной гипертензии и, в последующем, к легочному сердцу.
Уменьшение объема легочной паренхимы встречается при пневмэктомии, деструктивных процессах в легких и также приводит к рестриктивному нарушению вентиляции легких.
Отек легких. Отек легких – это избыточное накопление жидкости сосудистого происхождения в интерстиции легочной паренхимы или в полости альвеол. В норме существует равновесие между выходом в интерстиций фильтрата крови и её удалением с лимфой (лимфатический дренаж). Отек представляет собой преобладание фильтрации над дренажем вследствие первичного увеличения выхода жидкости из сосудов, либо нарушения лимфатического дренажа при нормальной фильтрации. В самом начале процесса излишек жидкости накапливается в интерстции межальвеолярных перегородок (интерстициальный отек легких), а впоследствии жикость выходит в полость альвеол (альвеолярный отек легких). Оба этих процесса уменьшают объем альвеолярного пространства и нарушают легочную диффузию.
Причинами отека легких могут быть: а) факторы повышающие гидростатическое давление крови в сосудах малого круга (гидростатический фактор, кардиогенный отек легких); б) повышение проницаемости сосудистой стенки при вдыхании окислов азота, фосгена, при гппероксии, аспирации воды или желудочного сока, при действии эндотоксинов, лучевых поражениях (мембраногенный фактор, токсический отек легких); в этом случае интерстициальный отек легких наступает при нормальном давлении крови в капиллярях; в) блок лимфатического дренажа (лимогенный отек). В каждом случае патогенез отека легких зависит от вызвавшей его причины.
В легких существуют различные противоотечные механизмы. Так, проницаемость для воды альвеолярного эпителия меньше, чем проницаемость сосудистого эндотелия, что препятствует, какое-то время, выходу жидкости из интерстиция в альвеолы. Более того, повышение давления жидкости в интерстиции приводит к её обратной фильтрации в сосуды, что препятствует развитию альвеолярного отека. Накопление жидкости в интерстиции разбавляет белки, уменьшает онкотическое давление, что также способствует возврату жидкости в просвет сосудов. Существенным компенсаторным противоотечным механизмом является усиление лимфатического дренажа и отток избытка интерстициальной жидкости.
Гиперемия легких (артериальная и венозная гиперемия) имеет в качестве главного патогенетического звена увеличение давления крови в легочных капиллярах и венах с нарушением циркуляции в малом круге кровообращения и в бронхиальных сосудах большого круга кровообращения.
Увеличение давления крови в бассейне легочных артерий, в капиллярах и легочных венах увеличивают фильтрацию жидкости из сосудов в межклеточное пространство и в альвеолы (транссудация, межклеточный и альвеолярный отек). Отек, в свою очередь, уменьшает податливость и растяжимость альвеол, увеличивает сопротивление, оказываемое диффузии газов, увеличивает альвеолярное мертвое пространство (альвеолы, которые не участвуют в диффузии), увеличивают количество неоксигенированной крови притекающей к левому сердцу – венозная примесь (уменьшается процент оксигемоглобина в артериальной крови), приводит к гипоксемии и гиперкапнии. В хронических случаях происходит перерождение стенок кровеносных сосудов и альвеол – возникает пневмосклероз, зарастание сосудов соединительной тканью, уменьшение емкости малого круга, гипертензия в малом круге, гиперфункция, гипертрофия и недостаточность правого желудочка.
Застой крови в бронхиальных венах ведет к набуханию слизистой бронхов, сужению их просвета и увеличению аэродинамического сопротивления.
Особым случаем легочного застоя и отека легких является острая недостаточность левого желудочка – сердечная астма.
Легочный застой проявляется одышкой, гипервентиляцией, рестриктивными и обструктивными нарушениями вентиляции, нарушением диффузии газов.
Острый респираторный дисстресс-синдром у взрослых. Острый респираторный дисстресс-синдром («шоковое легкое», болезнь гиалиновых пленок) представляет собой симптомокомплекс, который включает воспаление и инфильтрацию легочной паренхимы, увеличение проницаемости альвеоло-капиллярного барьера, альвеолярный легочный отек, формирование протеиновых пленок, которые покрывают альвеолярную поверхность. Летальность при этом синдроме составляет ок. 50%. Причинами острого респираторного дисстресс-синдрома является диссеминированное внутрисосудистое свертывание крови, ожоги, обширные травмы, гемморрагический шок, вдыхание жидкостей (например, при утоплении), тотальные пневмонии, массивные трансфузии, обширные микроэмболии, внутрисосудистая агрегация клеток крови, инактивация альвеолярного сурфактанта. Результатом действия этих факторов является значительное увеличение проницаемости биологических мембран, включая и альвеолярно-капиллярный барьер, усиленная фильтрация и накопление в альвеолах внутрисосудистой жидкости богатой протеинами, включая фибриноген. Коагуляция протеинов формируют гиалиновые пленки, которые покрывают альвеолы и препятствует диффузии газов, что приводит к тяжелой гипоксемии, которая не устраняется даже при вдыхании чистого кислорода. Индурация стенок альвеол уменьшает податливость легких, а инактивация сурфактанта ведет к их спадению и формированию множественных микроателектазов.
Острый респираторный дистресс-синдром у новорожденных. Острый респираторный дистресс у новорожденных имеет в своей основе два патогенетических фактора: ишемия легочной паренхимы и недостаточность образования альвеолярного сурфактанта.
Ишемия альвеолярной паренхимы с гипоксией ведет к увеличению проницаемости биологических мембран и обильной фильтрации внутрисосудистой жидкости в межклеточное пространство и в альвеолы. Белки, которые входят в состав плазмы крови, включая фибриноген, формируют «гиалиновые» пленки, которые покрывают поверхность альвеол.
Альвеолярный сурфактат синтезируется у плода, начиная с 20-ой недели пренатального периода, но более активно – после 35–36-ой недели. Это объясняет большую частоту случаев острого респираторного дистресс синдрома у недоношенных детей. До рождения объем легких ребенка составляет ок. 40 мл, а при переходе на внешнее дыхание – ок. 200 мл. Первый вдох у здорового новорожденного происходит без участия сурфактанта и, для разлипания альвеол, нуждается в большом транспульмонарном давлении, равном 40 ммм рт. ст. После разлипания альвеол, произведенного первым вдохом, вступает в действие альвеолярный сурфактант, который уменьшает поверхностное натяжение альвеол, снижает необходимое усилие для их расправлнения при вдохе и, таким образом, облегчает последующие дыхательные движения. При недостаточности сурфактанта поверхностное натяжение альвеол большое, сопротивление альвеол при их расширении так же большое, что требует значительных усилий дыхательной мускулатуры не только для осуществления первого, но и всех последующих вдохов. У этих детей, после первого вдоха, амплитуда дыхания прогрессивно уменьшается, несмотря на сильные сокращения дыхательных мышц. Создается впечатление, что мышцы не в состоянии раскрыть ригидные легкие. В зависимости от тяжести, процесс длится 4–5 дней, а максимальная летальность наблюдается на протяжении первых двух дней жизни ребенка.
Образование гиалиновых пленок на поверхности альвеол нарушает альвеолярно-капиллярную диффузию, вызывая гипоксемию.
Заключение. Одним из следствий первичных повреждений легочной паренхимы является внутрипаренхимальная легочная рестрикция – уменьшение дыхательного объема пропорционально уменьшению объема легких, нарушения соотношения вентиляции и перфузии легких, внутрилегочное шунтирование, нарушение диффузии кислорода, умеренная гипоксемия в покое и тяжелая гипоксемия при физической нагрузке, дыхательная недостаточность рестриктивного типа. В ответ на гипоксемию появляется легочная гипервентиляция, направленная на поддержание минутного объема дыхания посредством увеличения частоты дыхания.
Более поздними последствиями являются воспаление и замещение паренхимы легких фиброзной тканью, сокращение количества кровеносных сосудов в легких (уменьшение васкуляризции) одновременно с увеличением периферического сопротивления в малом круге кровообращения, легочная гипертензия, легочное сердце.
Рестрикция легких любого генеза приводит к рестриктивной дыхательной недостаточности. Рестриктивные повреждения характеризуются уменьшением объемов легких: общего объема, жизненной емкости, дыхательного объема и остаточного функционального объема легких, одновременно с сохранением нормального сопротивления воздухоносных путей. Клинически, рестриктивная недостаточность проявляется гипоксемией, которая резко нарастает при физических нагрузках.
34.1.1.5. Обструкция верхних дыхательных путей
Воздухоносные пути служат для проведения атмосферного воздуха к альвеолам и составляют воздухоносную систему (лишь в дыхательных бронхиолах имеет место обмен газов). Воздухоносные пути состоят из трахеи, главных, лобарных, сементарных бронхов, терминальных бронхиол и, частично, дыхательных бронхиол. Последние делятся на 2–11 альвеолярных протоков, которые переходят в альвеолярные мешочки, состоящие из альвеол – функциональная единица, где происходит обмен газов.
Стенки воздухоносных путей, вплоть до бронхов с диаметором до 1 мм, защищены от спадения хрящевым каркасом. Все структуры воздухоносной системы снабжены гладкой мускулатурой, и лишь альвеолы не обладают сократимостью. Терминальные и дыхательные бронхиолы также наделены гладкими мышцами, однако в их стенке отсутствует механическая хрящевая подложка, что делает возможным их спазмирование до полного закрытия просвета, как это бывает при приступах бронхиальной астмы.
Воздухоносные пути обладают аэродинамическим механическим сопротивлением. Учитывая тот факт, что движение воздуха через воздухоносные пути имеет преимущественно ламинарный характер (лишь в местах разветвления, сужения или расширения движение становится турбулентным), аэродинамическое сопротивление может быть описано уравнением Hagen – Poiseuille:
R= 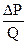 ,
,
где ΔР – это разница между атмосферным и внутриальвеолярным давлением, Q – объемная скорость вдыхаемого воздуха, R – аэродинамическое сопротивление. Аэродинамическое сопротивление зависит от плотности вдыхаемого воздуха: например, сжатый воздух имеет большую плотность и, поэтому, обладает большим сопротивлением, чем воздух при нормальном атмосфернмм давлении. Таким образом, аэродинамическое сопротивление дыхательных путей является изменчивой величиной, которая зависит от диаметра воздухоносных путей (увеличивается при сужении бронхов), от плотности воздуха (увеличивается параллельно увеличению давления), от характера движения воздуха (растет при переходе от ламинарного движения к турбулентному), от объемной скорости воздуха (увеличивается пропорционально увеличению скорости). Все это определяет то, что при спокойном дыхании, аэродинамическое сопротивление дыхательных путей меньше, чем эластическая сила легких и выдох осуществляется пассивно. При форсированном, учащенном дыхании, аэродинамическое сопротивление превышает эластическую силу легких, из-за чего необходима дополнительная энергии для выполнения выдоха, который становится активным. Аэродинамическое сопротивление вместе с эластическим сопротивлением альвеол и неэластическим сопротивлением тканей грудной клетки, определяют дыхательное усилие – механическую работу, выполняемую дыхательной мускулатурой.
Наиболее выраженной формой поражения функции дыхательных путей является их обструкция.
Обструкцией называют увеличение сопротивления воздухоносных путей, которое препятствует или делает невозможной легочную вентиляцию и приводит к обструктивной недостаточности дыхания. Обструкция дыхательных путей делится по анатомической локализации, степени сужения (стеноза) путей и по биомеханике дыхания:
1) обструкция, которая нарушает одновременно вдох и выдох:
а) сжатие или сдавление верхних дыхательных путей;
б) спазм с обструкцией мелких дыхательных путей (хронический обструктивный бронхит, бронхиальная астма);
2) лабильная обструкция, которая зависит от фазы и особенностей дыхания (форсированный вдох и выдох):
а) обструкция, преимущественно на выдохе (паралич голосовых связок, размягчение трахеи в части расположенной вне грудной клетки);
б) обструкция преимущественно на выдохе (коллапс трахеи при размягчении трахеи в части расположенной в грудной клетке, бронхиальный коллапс или коллапс бронхиол при эмфиземе легких).
При обструкции воздухоносных путей сопротивление потоку воздуха увеличивается на величину, равную кубу радиуса, что вызывает значительное затруднение дыхания. Так, при уменьшении радиуса бронхов в 2 раза, сопротивление возрастает в 16 раз. По этой причине, даже незначительное уменьшение просвета воздухоносных путей существенно увеличивает сопротивление. В этом отношении особую опасность представляют отделы воздухоносных путей проксимальнее бифуркации трахеи, которые обусловливают примерно 80% общего сопротивления бронхиального древа.
Обструкция гортани или трахеи (инородные тела, опухоли, отек) ведет к летальным нарушениям вентиляции – асфиксии.
Асфиксия представляет собой острую дыхательную недостаточность, характеризующуюся одновременным нарушением поступления кислорода (гипоксемия) и выделения углекислого газа (гиперкапния). В развитии асфиксии выделяют несколько периодов. Первый период проявляется частым и глубоким дыханием с преимущественным затруднением вдоха – инспираторная одышка. Второй период характеризуется прогрессивным уменьшением частоты дыхания с сохранением максимальной амплитуды и экспираторной одышкой. В третьем периоде, одновременно с уменьшением частоты, уменьшается и амплитуда дыхания; этот период постепенно приводит к остановке дыхания (терминальная пауза), за которой следует восстановление дыхания на короткий период (агональное, терминальное дыхание, гаспинг-дыхание), завершающейся окончательной остановкой дыхания – клиническая смерть.
При обструкции бронхов большого калибра (например, внутрибронхиальный рост опухоли) вентиляция данной области легкого (доля, сегмент) отсутствует, заключенный в этой области воздух рассасывается, а легкие спадаются – происходит обструктивный ателектаз.
34.1.1.5 Обструкция нижних воздухоносных путей
Обструкция бронхиол – главное патогенетическое звено бронхиальной астмы и хронического обструктивного бронхита. Характеризуется сужением мелких дыхательных путей (метасегментарных бронхов и терминальных бронхиол). Сужение вызвано их спазмом, накоплением слизи, воспалением и набуханием слизистой. Плюс к этому, выдох сопровождается дополнительной обструкцией, патогенез которой состоит в том, что эти мелкие воздухоносные пути лишены хрящевой подложки и, по этой причине, высокое давление, которое создается в легких во время выдоха, сжимает их до полного коллапса. Эту же роль играет и капелька слизи, находящаяся в просвете бронхиол, которая ведет себя подобно клапану - во время вдоха переносится по направлению к альвеолам, что не препятствует вдоху, однако во время выдоха отступает в бронхиолу, которую закупоривает, препятствуя выдоху. Любое хроническое нарушение выдоха приводит к гиповентиляции легких и увеличению остаточного объема – возникает острая легочная эмфизема.
В конечном итоге, обструктивная недостаточность дыхания характеризуется увеличением сопротивления воздухоносных путей на вдохе или на выдохе, инспираторной либо экспираторной одышкой, уменьшением резервов вдоха и выдоха, увеличением функциональной остаточной емкости, альвеолярной гиповентиляцией, сдавлением слабо вентилируемых легочных областей, вазоконстрикцией, увеличением сосудистого сопротивления в невентилируемых областях.
Бронхиальная астма или гиперреактивность воздухоносных путей представляет собой их хроническое воспаление с патогенетическим участием различных клеток мезенхимального происхождения - мастоцитов, эозинофилов, Т-лимфоцитов, макрофагов, нейтрофилов и эпителиальных клеток. У предрасположенных к астме людей воспаление вызывает повторные приступы одышки, затрудненное дыхание, кашель, особенно ночью или утром.
Патогенез астмы является комплексным и включает 3 главных компонента: воспаление воздухоносных путей, перемежающаяся обструкция и гиперчувствительность бронхов.
Воспаление воздухоносных путей при бронхиальной астме может быть острым, подострым и хроническим, но наличие отека или слизи способствует обструкции и гиперреактивности бронхов. Основные клетки, участвующие в воспалении воздухоносных путей выделяют медиаторы воспаления и аллергии. Мастоциты и эозинофилы секретируют гистамин, факторы хемотаксиса, лейкотриены, простагландины, катионные белки, макрофаги, активные Т-лимфоциты поддерживают воспалительный процесс посредством выделения цитокинов, фибробласты, эпитолиоциты, эндотелиальные клетки способствуют хроническому течению данного процесса. Такие факторы, как молекулы адгезии (селектины, интегрины) способствуют переходу воспалительного процесса на воздухоносные пути. В итоге, происходит инфильтрация стенок бронхиол мононуклеарными клетками, нейтрофилами и эозинофилами, гипертонус гладких мышц бронхов, гиперсекреция слизи, отшелушивание эпителия, гиперплазия гладких мышц и ремоделирование воздухоносных путей.
Обструкция воздухоносных путей при бронхиальной астме вызвана сужением просвета бронхов, отеком, образованием слизистых пробок, ремоделированием воздухоносных путей (деформация, утолщение, сужение). Степень обратимости обструкции зависит от структурных изменений дыхательных путей, вызванных воспалением.
Гиперреактивность воздухоносных путей обусловливает чрезмерный спастический ответ бронхов на многочисленные неспецифические стимулы (на температуру и влажность вдыхаемого воздуха, на загрязнение воздуха, на физическую работу, на психогенные факторы). Как правило, клиническая тяжесть астмы коррелирует со степенью гиперчувствительности бронхов.
Существует также и астма (правильнее, бронхоспазм), провоцируемая физическим усилием, патогенез которой является противоречивым. Физическое напряжение действует в качестве триггера, запускающего острый спазм бронхов с повышенной реактивностью. Встречается такой вид астмы у лиц, страдающих атопией, аллергическим ринитом, фиброкистозом и даже у здоровых людей. Клиницисты часто игнорируют эту форму астмы. Болезнь, вероятно, опосредована потерей воды и тепла из воздухоносных путей. Так, оптимальными для дыхательных путей являются температура вдыхаемого воздуха равная 370С и относительная влажность воздуха равная 100%. При гипервентиляции, вызванной физической работой (или при эмоциональной гипервентиляции), носовые ходы не в состоянии обеспечить необходимый транзит воздуха, что вынуждает дышать ртом. При этом вдыхаемый воздух не увлажняется и не согреватеся, что и вызывает бронхоспазм. Бронхоальвеолярный смыв в этих случаях не демонстрирует повышения медиаторов воспаления.
Таким образом, при обструктивных повреждениях первоначально сохранен весь функциональный потенциал дыхательного аппарата (реализация дыхательного усилия, растяжимость и эластичность структур), за исключением воздухопроводящей способности легких – увеличивается сопротивление воздухоносных путей. В дальнейшем, после установления газового и кислотно-щелочного дисбаланса, нарушается и функция дыхательного центра с прогрессированием патологических процессов вплоть до торможения и остановки дыхания.
Общим последствием обструкции воздухоносных путей, как верхних, так и нижних, является обструктивная дыхательная недостаточность.
Дата добавления: 2014-12-23; просмотров: 1605; Мы поможем в написании вашей работы!; Нарушение авторских прав |