КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Билет №15
1. Реакции нуклеофильного замещения у sp2-гибридизированного атома углерода (карбоновые кислоты и их функциональные производные). Реакции ацилирования (образование ангидридов, сложных эфиров, сложных тиоэфиров, амидов) и обратные им реакции гидролиза. Сравнительная активность ацилирующих реагентов (ангидридов, карбоновых кислот, сложных эфиров, сложных тиоэфиров). Ацилфосфаты и ацилкофермент А. Биологическая роль реакций ацилирования.
Реакции нуклеофильного замещения характерны для карбоновых кислот и их функциональных производных. Они обусловлены способностью заместителя Х в соединениях типа R-CO-X уходить в виде аниона Х- или сопряженной кислоты НХ. Их результатом является замещение Х на другую нуклеофильную группу Y. Общая схема механизма таких реакций включает образование нестабильного продукта присоединения нуклеофила к атому углерода карбонильной группы.
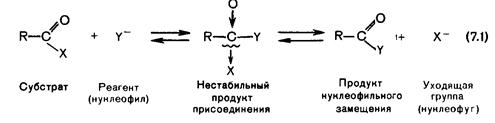
Реакция протекает либо при наличии достаточно сильного нуклеофила Y- и хорошо уходящей группы Х, либо в условиях кислотного катализа. Последний часто оказывается необходимым, так как вследствие +М-эффекта заместителя Х частичный положительный заряд на карбонильном атоме углерода оказывается недостаточным для атаки его нуклеофилом. В таких случаях протонирование кислорода карбонильной группы ведет к появлению полного положительного заряда на атоме углерода, что облегчает атаку нуклеофилом. Большая часть этих реакций ацилирования, т.е. введения ацильной группы R-CO в органическую молекулу вместо водорода. Галогенангидриды (R-CO-Hal) обычно получают действием на карбоновые кислоты или их соли галогенидов фосфора или серы, чаще всего используют хлорид фосфора (V) PCl5, хлорид фосфора (III) PCl3 и оксид-дихлорид серы (тионилхлорид) SOCl2. Последний особенно удобен, так как, кроме целевого хлорангидрида, дает только газообразные продукты.

Галогенангидриды – наиболее активные ацилирующие реагенты среди производных карбоновых кислот. Так, ацетилхлорид легко гидролизуется водой (ацилирует воду) с выделением тепла и образованием уксусной кислоты. Ангидриды карбоновых кислот могут содержать остатки одинаковых кислот R – CO – O – CO – R и могут быть смешанными R – CO – O – CO – R’. Для получения простых ангидридов кислоты нагревают в присутствии водоотнимающих средств, например оксида фосфора (V) P2O5, или обрабатывают соль кислоты галогенангидридом. Смешанные ангидриды можно приготовить только вторым способом.
Реакция этерификации в отсутствие катализаторов протекает чрезвычайно медленно вследствие низкой активности карбонильной группы. Однако в присутсвии минеральных кислот (серной, хлороводородной) реакция существенно ускоряется.
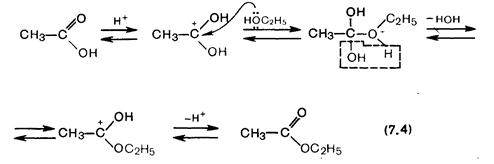
Процесс этерификации обратим. Сдвиг равновесия вправо осуществляется за счет удаления из реакционной смеси одного из конечных продуктов.

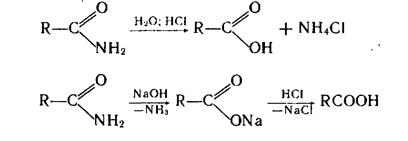
Амиды могут быть первичными, вторичными и третичными. За счет взаимодействия неподелённой пары электронов атома азота с π-электронами поляризованной двойной связи карбонильной группы связь C-N в амидах имеет частично двойной характер. Поэтому свободное вращение вокруг данной связи заторможено и амидная группировка имеет строение, близкое к плоскому, что важно для структуры белков. Другим следствие +М-эффекта аминогруппы является понижение основности амидов по сравнению с аминами. Амиды дают непрочные, легко гидролизирующиеся соли только с очень сильными кислотами. Амиды проявляют амфотерные свойства. Они могут реагировать с сильными основаниями, например амидом натрия, образую соответствующие соли.

По сравнению с другими производными карбоновых кислот амиды наименее склонны подвергаться нуклеофильному замещению. Это справедливо и в отношении их гидролиза, который осуществляют в кислой или щелочной среде в достаточно жестких условиях.
Сложные тиоэфиры — органические соединения, содержащие функциональную группу C-S-CO-C и являющиеся сложными эфирами тиолов и карбоновых кислот. Сложные тиоэфиры играют важную роль в биохимических процессах, наиболее известный представитель этого класса — ацетил-CoA. Способы получения сложных тиоэфиров разнообразны, но важнейшим является конденсация тиолов и карбоновых кислот в присутствии водоотнимающих реагентов.
RSH + R’COОH → RSC(O)R' + H2O
Ангидриды карбоновых кислот и некоторые лактоны реагируют подобным же образом с тиолами в присутствии оснований.
Гидролиз сложных эфиров может быть осуществлен как в кислой, так и в щелочной среде. Гидролиз в щелочной среде необратим и требует эквимолекулярных количеств щелочи. Приведенная ниже схема механизма справедлива для щелочного гидролиза не только сложных эфиров, но также тиоэфиров, галогенангидридов, ангидридов и амидов карбоновых кислот. Причиной необратимости гидролиза в щелочной среде является образование стабильного ацилат-иона.
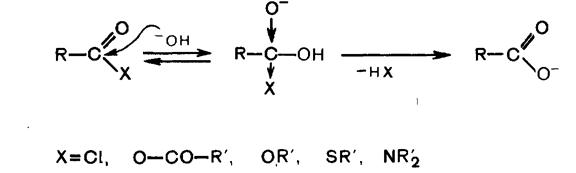
Легкость гидролиза зависит от эффективного положительного заряда на карбонильном углероде соединения R-CO-X, величина которого определяется природой Х, т.е. совместным действием его –I и +M-эффектов.
Реакционная способность ацильных соединений определяется как величиной положительного заряда на атоме углерода карбонильной группы, так и способностью уходящей группы уходить.
Величина положительного заряда Сd+=Оd- группы и, следовательно, активность реагента увеличивается с повышением электроноакцепторных свойств радикала. Так, константа диссоциации и ацилирующая активность кислот увеличивается в ряду:
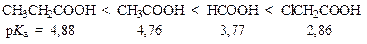
В ацильных соединениях, полученных из одной и той же кислоты, величина d+ является результатом взаимодействия электронных облаков карбонильной и уходящей групп:

Она увеличивается при возрастании отрицательного индукционного эффекта и уменьшении положительного эффекта сопряжения.
Способность группы Y уходить зависит от того, каким основанием она является: чем сильнее основание, тем хуже уходит. При определении силы основания обычно используют константу диссоциации сопряженной с ним кислоты: чем сильнее кислота, тем слабее сопряженное с ней основание:

В связи с этим ацилирующая активность производных карбоновой кислоты уменьшается от хлорангидрида к амиду.
Ацилфосфаты являются макроэргическими фосфатами карбоновых кислот. У них остаток фосфорной кислоты соединяется макроэргической связью с кислородом карбоксильной группы кислоты.
Важнейшие представители ацилфосфатов – 1.3-дифосфоглицериновая кислота, ацетилфосфат, сукцинилфосфат и др. 1,3-дифосфоглицериновая кислота образуется в качестве промежуточного продукта в реакциях цикла Кальвина и анаэробной стадии дыхания, где этот макроэргический фосфат участвует в синтезе АТФ. Ацетилфосфат является промежуточным продуктом в синтезе ацетилкофермента А, принимающего участие во многих биосинтетических реакциях, а сукцинилфосфат – промежуточным метаболитом при фосфоролизе сукцинилкофермента А, который происходит в цикле Кребса и служит источником образования АТФ (у растений) или ГТФ (у человека и животных).
Ацилирование — замещение в молекуле органического соединения атома водорода или металла ацилом, т.е. остатком молекулы органической кислоты. Частным случаем ацилирования является ацетилирование — присоединение остатка уксусной кислоты СН3СО—. В организме реакции ацилирования протекают с участием особого вещества — кофермента ацилирования КоА (KoA-SH). Кислота вначале активируется путем соединения с сульфгидрильной группой KoA-SH (образуется ацил-S-KoA, или ацилкофермент А). Эта реакция протекает с использованием энергии макроэргической связи аденозинтрифосфорной кислоты (АТФ). Остаток кислоты затем переносится с ацил-S-KoA на ацилируемое вещество. Таким путем происходит, например, присоединение остатков жирных кислот (стеариновой и др.) к глицеринфосфорной кислоте в процессе синтеза жиров: 1) KoA-SH + стеариновая кислота + АТФ → стеарил-S-KoA + АДФ + пирофосфорная кислота; 2) стеарил-S-KoA + глицеринфосфорная кислота → стеарилглицеринфосфорная кислота + KoA-SH.
Важнейшее значение в процессах обмена веществ имеет активная форма уксусной кислоты — ацетил-S-KoA, которая образуется при аэробном распаде углеводов, аминокислот, жирных кислот. С другой стороны, ацетил-S-KoA участвует в синтезе разнообразных веществ, в том числе жирных кислот и аминокислот. Таким образом, ацетил-S-KoA оказывается звеном, в котором перекрещиваются пути обмена основных компонентов организма. Кроме того, остаток уксусной кислоты ацетил-S-KoA может быть окислен до CO2 и H2O в цикле трикарбоновых кислот, причем энергия этого окислительного процесса аккумулируется в АТФ. Две молекулы ацетил-S-KoA могут взаимодействовать с образованием ацетоацетил-S-KoA; это соединение распадается на ацетоуксусную кислоту и KoA-SH. Существенна также роль ацилирования в процессах обезвреживания и выделения ядовитых или не свойственных организму веществ. Бензойная кислота обезвреживается в печени путем образования гиппуровой кислоты. Вначале образуется бензоил-S-KoA, который ацилирует глицин с образованием гиппуровой кислоты. На этом свойстве печени основана функциональная проба Квика. Многие лекарственные вещества (сульфаниламиды, фтивазид, ПАСК и др.) в значительной части выводятся из организма в виде ацетильных производных.
К реакциям биологического ацилирования относится также и биосинтез белков. В этой реакции не участвует КоА: аминокислота активируется по карбоксильной группе путем образования ангидридной связи с адениловой кислотой. Активированная аминокислота ацилирует другую аминокислоту по аминогруппе, в результате чего образуется пептидная связь.
2. Лекарственные средства на основе модифицированных нуклеиновых оснований (фторурацил, меркаптопурин). Нуклеозиды-антибиотики. Принцип химического подобия. Изменение структуры нуклеиновых кислот под действием химических веществ. Мутагенное действие азотистой кислоты.
В качестве лекарственных средств в онкологии используют синтетические производные пиримидинового и пуринового рядов, по строению похожие на естественные метаболиты (в данном случае – на нуклеиновые основания), но не полностью им идентичные, т.е. являющиеся антиметаболитами. Например, 5-фторурацил выступает в роли антагониста урацила и тимина, 6-меркаптопурин – аденина. Конкурируя с метаболитами, они нарушают синтез нуклеиновых кислот в организме на разных этапах.
В клетках в свободном состоянии содержатся некоторые нуклеотизы, не являющиеся компонентами нуклеиновых кислот. Эти нуклеозиды обладают антибиотической активностью и приобретают всё большее значение при лечении злокачественных образований. Они отличаются от обычных нуклеозидов некоторыми деталями строения либо углеводной части, либо гетероциклического основания. Это позволяет им выступать, по-видимому, в роли антиметаболитов. Нуклеозидные антибиотики пиримидинового ряда часто подобны цитидину, пуринового ряда – аденозину. «Небольшой» разницы в строении или конфигурации всего лишь одного атома углерода в углеводном остатке зачастую достаточно, чтобы соединение выполняло роль ингибитора биосинтеза (белка, ДНК) и размножения (вирусов, грибков, бактерий).
Одной из причин возникновения мутаций служит воздействие химических факторов. Наиболее распространённый вид мутаций – замена какой-либо пары оснований на другую, одной из причин которого может являться сдвиг таутомерного равновесия. Например, тимин в лактамной форму не образует водородные связи с гуанином, а в лактимной – образует, что приводит к замене обычной пары тимин-аденин на пару тимин-гуанин.
Если на аденозин подействовать азотистой кислотой, то в результате реакции дезаминирования аминогруппа превратится в гидроксильную, вследствие чего из аденозина получается другой нуклеотид – инозин, содержащий гипоксантин. Это может привести к замене в ДНК комплементарной пары оснований, так как адениновый нуклеотид комплементарен тиминовому, а инозин может образовывать комплементарную пару только с цитидиновым нуклеозидом.

3. Приведите проекционную формулу стереоизомера природной α-аминокислоты, содержащей в радикале гидрокси-группу, в форме катиона.
Серин:
HO – CH2 – C*H – COOH
|
NH2
Природный стереоизомер (L-форма) D-форма
COОH COОH
 | |

 H3N+ Н Н NH3+
H3N+ Н Н NH3+
CH2OH CH2OH
4. Напишите строение участка одной цепи ДНК, включая три разных нуклеотида.
Наже приведена структура участка одной цепи ДНК, включающая в себя четыре разных нуклеотида (рядом с нуклеотидами латиницей указаны сокращения названий этих нуклеотидов).

5. Как химическим путём различить растворы глюкозы и этилглюкозида?
 В молекуле глюкозы одна из гидроксильных групп существенно отличается по свойствам от всех остальных (см. реакцию ниже).
В молекуле глюкозы одна из гидроксильных групп существенно отличается по свойствам от всех остальных (см. реакцию ниже).
Такая реакция не характерна для этилглюкозида, водород этой гидроксогруппы уже замещён на этильный радикал.
Дата добавления: 2015-02-10; просмотров: 3033; Мы поможем в написании вашей работы!; Нарушение авторских прав |