КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Болезнь Брутона
ГЛАВА 3. ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
Определение, клинические проявления и классификация иммунодефицитных состояний. Классификации первичных ИДС. Первичные ИДС адаптивного иммунитета с преимущественным поражением В-клеточного звена. Первичные ИДС адаптивного иммунитета с преимущественным поражением Т-клеточного звена. Комбинированные Т- и В-иммунодефициты. Первичные дефициты фагоцитарного звена врожденного иммунитета. Первичные дефициты системы комплемента. Предварительная диагностика первичных ИДС. Определение, основные причины формирования вторичных иммунодефицитов. Физиологические (возрастные) иммунодефициты. Роль предшествующих заболеваний, ятрогенных факторов, неблагоприятных экологических факторов и факторов риска образа жизни в возникновении вторичных ИДС.
В функционировании иммунной системы, как и любой другой системы организма, могут возникать нарушения, которые ведут к развитию заболеваний, характерных прежде всего для этой системы. Одним из самых распространенных вариантов таких нарушений являются иммунодефицитные состояния (ИДС), характеризующиеся неспособностью иммунной системы развивать нормальный иммунный ответ.
Иммунодефицитные состояния (ИДС) – это снижение функциональной активности основных компонентов иммунной системы, ведущее к нарушению иммунологического реагирования, к снижению или к формированию неадекватного врожденного или адаптивного иммунного ответа на самые различные патогены и антигены, в первую очередь – на инфекционные.
Необходимо отличать понятие иммунодефицит от понятия иммунологической толерантности, при которой также снижен или отсутствует иммунный ответ. В отличие от иммунодефицита, иммунологическая толерантность всегда специфична, то есть она формируется только против одного конкретного антигена или группы антигенов. При иммунодефицитных состояниях, как правило, снижается иммунологическая реактивность в целом.
Наличие ИДС в первую очередь приводит к снижению противоинфекционной защиты организма, что клинически проявляется в повышении инфекционной заболеваемости.
Для ИДС с преимущественным поражением В-клеточного звена адаптивного иммунитета характерно возникновение рецидивирующих инфекций, в том числе вызванных стафилококками, стрептококками, пневмококками, возбудителями кишечных инфекций и др.
При ИДС с преимущественным поражением Т-клеточного звена адаптивного иммунитета в дополнение ко многим бактериальным инфекциям возникают инфекционные заболевания, вызванные такой условно-патогенной флорой, как грибы Candida, вирусы (герпес, цитомегаловирус, аденовирусы), пневмоцисты, внутриклеточные бактерии (микобактерии, уреаплазмы и др.). Характерна также склонность к генерализации туберкулезной инфекции, повышена частота гельминтозов.
Первичные ИДС могут быть также обусловлены дефектами компонентов врожденного иммунитета. Эти дефекты могут затрагивать самые разные звенья системы врожденного иммунитета: компоненты и ингибиторы системы комплемента, дефекты фагоцитарных клеток, NK-клеток, рецепторов и транспортных молекул (ТАР), участвующих в процессинге антигенов. В связи с интенсивными исследованиями в этой области, которые проводятся с применением современных генетических и молекулярно-биологических методов, выявляются все новые варианты первичных ИДС врожденного иммунитета, а также сочетанных ИДС врожденного и адаптивного иммунитета. Как правило, первичные ИДС врожденного иммунитета проявляются в виде самых различных инфекционных заболеваний, например:
· Дефицит компонентов комплемента (С1, С4, С2; С3, С5-9 и др.) приводит к рецидивирующим генерализованным инфекциям, вызванным инкапсулированными микроорганизмами (Neisseria meningitidis, пневмококк, гемофильная палочка). Часто встречается системная красная волчанка.
· Дефекты фагоцитарной системы клинически чаще проявляются в виде инфекционных поражений кожи и паренхиматозных органов, например, вызываемых стафилококками и клебсиеллами при хронической гранулематозной болезни.
К клиническим проявлениям ИДС относятся также гематологические нарушения (лимфоаденопатии, лейкозы), характерно возникновение поражений желудочно-кишечного тракта, обусловленных в значительной мере нарушением местного иммунитета ЖКТ. Часто ИДС сопровождаются возникновением аутоиммунных и аллергических заболеваний. Особое место принадлежит онкологическим заболеваниям. Чаще всего у больных с ИДС встречаются гемобластозы, лимфопролиферативные заболевания, саркома Капоши.
Все иммунодефициты принято разделять на первичные, или врожденные, и вторичные, или приобретенные. Вторичные ИДС не являются генетически детерминированными, такие дефекты иммунной системы приобретаются в течение жизни и являются следствием каких-либо повреждающих воздействий на иммунную систему или наличия других заболеваний.
Первичные иммунодефицитные состояния
Первичные ИДС – это наследственные нарушения врожденной и/или адаптивной иммунной системы, связанные с генетическими дефектами одного или нескольких компонентов. Первичные ИДС адаптивной иммунной системы обусловлены преимущественными дефектами ее В-клеточного и Т-клеточного звена, во многих случаях имеют место сочетанные поражения обоих звеньев. Наиболее изученными первичными ИДС врожденного иммунитета являются ИДС, обусловленные генетическими дефектами системы комплемента и фагоцитарной системы.
При нормальном течении беременности во внутриутробном периоде развития ребенок находится в стерильных условиях. Немедленно после рождения его организм начинает заселяться микроорганизмами. Так как окружающая человека микрофлора не является патогенной, эта колонизация не вызывает болезни. В последующем встреча с патогенными микроорганизмами, с которыми ребенок еще не встречался, вызывает развитие соответствующей инфекционной болезни. Каждый контакт с патогеном приводит к расширению иммунологической памяти и формирует долговременный иммунитет.
В защите организма от постоянных атак патогенных вирусов, бактерий, грибов и простейших участвуют два основных компонента врожденной иммунной защиты: фагоцитоз и комплемент, а также два компонента адаптивной защиты: В-клеточный (антитела) и Т-клеточный. Каждый из этих компонентов может функционировать независимо, но чаще всего они работают в тесном комплексном взаимодействии. Врожденные дефекты одного из указанных звеньев приводят к нарушению защиты организма в целом и клинически проявляются в виде различных вариантов первичных иммунодефицитов.
В настоящее время, благодаря идентификации молекулярных нарушений, лежащих в основе патогенеза многих первичных иммунодфицитов, и в связи с большой вариабельностью клинической картины стало ясно, что этот вид патологии гораздо более распространен, чем считалось ранее.
Ранняя диагностика и адекватное лечение являются условием жизни пациентов с многими формами первичных иммунодефицитов. Без своевременной правильной диагностики и адекватного лечения ребенок погибает, как правило, в течение первого года жизни. Каждый вид иммунодефицита имеет свою, уже отработанную тактику лечения, и большинство первичных дефектов иммунной системы в настоящее время подлежит коррекции. Своевременная диагностика и лечение первичных иммунодефицитов являются одним из неиспользованных резервов для снижения заболеваемости, смертности и инвалидности населения в целом.
Первичные ИДС, как правило, имеют наследственную природу, поэтому их еще называют генетически обусловленными ИДС. Чаще всего первичные ИДС наследуются по рецессивному типу. В тех случаях, когда генетические дефекты затрагивают специфические факторы иммунитета (антителообразование, клеточные формы адаптивного иммунного ответа), их называют еще специфическими ИДС, в отличие от дефектов неспецифических (врожденных) компонентов защиты (фагоцитоза, системы комплемента и др.).
Иногда по отношению к первичным ИДС применяют термин «врожденные», что подразумевает возможность возникновения заболевания на основе наследственных дефектов, возникших в эмбриональном периоде, например, селективный дефицит IgA как следствие внутриутробно перенесенной коревой краснухи.
Первичные иммунодефициты разнообразны, и их описание и классификация в литературе постоянно меняются. Одной из первых классификаций иммунодефицитов адаптивной иммунной системы является предложенная в 1974 году Ю.М. Лопухиным и Р.В. Петровым классификация, в основу которой положены уровни генетических блоков на различных этапах дифференцировки Т- и В-лимфоцитов.
Согласно этой классификации, все многообразие форм ИДС адаптивной иммунной системы разделено на три группы:
1. Первичные ИДС с преимущественным поражением Т-системы (блок II, VI).
2. Первичные ИДС с преимущественным поражением B-системы (блок III, IV, V).
3. Комбинированные первичные ИДС с одновременным поражением Т-и В-систем (блок I, блоки V+VI и др.).
 |  |
|
|
 VI
VI

 |
 II Внутритимусные Периферические
II Внутритимусные Периферические
Т-лимфоциты Т-лимфоциты
 |
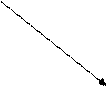
 I
I
 Стволовая III
Стволовая III
кроветворная
|
|



|
 |  | ||||||||
 | 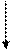 | 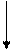 |
IgM IgG IgA
Рисунок 42. Патогенетическая схема классификации первичных ИДС Т- и В-систем иммунитета
(Р.В. Петров, Ю.М. Лопухин)
Блок I. Практически отсутствуют стволовые кроветворные клетки. Характерна генерализованная аплазия кроветворной и лимфоидной ткани. Не образуются ни Т-, ни В-лимфоциты.
К этому блоку относили тяжелую комбинированную иммунологическую недостаточность (ТКИН), которая встречается в различных формах.
Блок II. Блок на ранних этапах развития внутритимусных Т-лимфоцитов. Полное выключение клеточного адаптивного иммунитета. У таких больных характерно возникновение тяжелых рецидивирующих вирусных инфекций, приводящих к гибели в раннем (до 1 года) возрасте; высока частота врожденных аномалий развития; в 100-1000 раз выше риск возникновения злокачественных опухолей; организм пациента не отторгает чужеродный трансплантат.
Этот блок представлен, в частности, аплазией или гипоплазией тимуса (синдром Ди Джорджи).
Блок III. Для этого блока характерна агаммаглобулинемия, сцепленная с Х-хромосомой (болеют мальчики). У таких детей антитела практически не вырабатываются, что приводит в первую очередь к частым, тяжело протекающим бактериальным инфекциям. Этот вариант иммунодефицита называется болезнью Брутона, он диагносцируется, как правило, во втором полугодии жизни, так как в течение первых месяцев организм ребенка защищен материнскими антителами, прошедшими через плаценту и содержащимися в материнском молоке.
Блок IV. Несколько снижено число В-лимфоцитов. Гипогаммаглобулинемия с макроглобулинемией, так как синтез IgM сохранен или даже повышен, тогда как уровень IgG и IgA резко снижен.
Блок V. Селективный дефицит IgA. Из-за отсутствия основного секреторного иммуноглобулина у таких детей развиваются в первую очередь инфекционные заболевания, связанные со слизистыми оболочками носоглотки, дыхательных путей, ЖКТ, урогенитального тракта.
Блок VI. Затрагивает процессы созревания и выхода Т-лимфоцитов в периферические лимфоидные органы и кровяное русло. Характерна сочетанная патология с преимущественным поражением функций Т-клеточного иммунитета.
В настоящее время идентифицировано более 70 врожденных дефектов как врожденной, так и адаптивной иммунной системы. Вероятно, по мере совершенствования методов молекулярной иммунодиагностики, их число будет расти. Эндогенные, как правило, генетически обусловленные дефекты одного из компонентов иммунной системы приводят к нарушению всей системы защиты организма и клинически выявляются как одна из форм первичного иммунодефицитного состояния. Так как при нормальном функционировании иммунной системы в комплексном иммунном ответе участвуют многие типы клеток и сотни молекул, в основе патогенеза клинических форм первичных ИДС лежат многочисленные варианты дефектов.
В последние годы пришло осознание того факта, что клинические проявления первичных ИДС могут наблюдаться не только у новорожденных, но и в более позднем возрасте. Это связано с тем, что дефект одного из звеньев иммунитета может какое-то время не проявляться клинически в виде повышенной инфекционной заболеваемости, так как все другие компоненты иммунитета сохранены и компенсируют этот дефект пока не истощены их резервные возможности.
Первичные ИДС – это относительно редкие заболевания, их частота в среднем составляет 1/25000 - 1/100000, хотя селективный дефицит IgA встречается гораздо чаще: 1/500 - 1/700 человек. По данным Европейского регистра первичных ИДС за 1997 год частота зарегистрированных первичных ИДС в среднем по Европе составляла 1/96000 населения, тогда как в отдельных странах она была существенно выше: 1/38000 (Великобритания); 1/12500 (Швейцария); 1/10000 (Швеция). Скорее всего такие различия обусловлены уровнем развития медицины в разных странах, и реальная частота первичных ИДС существенно выше публикуемых средних цифр.
В настоящее время общепринятой является следующая упрощенная классификация первичных ИДС:
I. Первичные ИДС с преимущественным поражением гуморального адаптивного иммунитета
1. Сцепленная с Х-хромосомой агамма(гипогамма) глобулинемия – болезнь Брутона.
2. Общая вариабельная иммунологическая недостаточность (ОВИН) – общая вариабельная гипогаммаглобулинемия.
3. Транзиторная гипогаммаглобулинемия у детей (медленный иммунологический старт).
4. Избирательный дефицит иммуноглобулинов (селективный дефицит IgA).
II. Первичные ИДС с преимущественным поражением Т-клеточного адаптивного иммунитета
1. Синдром Ди Джорджи (гипо-, аплазия тимуса).
2. Хронический слизисто-кожный кандидоз.
III. Комбинированные Т- и В-иммунодефициты адаптивного иммунитета
1. Тяжелая комбинированная иммунологическая недостаточность (ТКИН):
а) Х-сцепленная;
б) аутосомно-рецессивные.
2. Атаксия – телеангиоэктазия (синдром Луи-Барр).
3. Синдром Вискотта-Олдрича.
4. Иммунодефицит с повышенным уровнем IgM (сцепленный с Х-хромосомой).
5. Иммунодефицит с карликовостью.
IV. Дефициты фагоцитарной системы
1. Хронический лимфогранулематоз.
2. Синдром Чедиака-Хигасси.
3. Синдром гипер-IgE (синдром Джоба).
V. Дефициты системы комплемента
1. Первичные ИДС, обусловленные дефицитом компонентов комплемента.
2. Первичные ИДС, обусловленные дефицитом инактиваторов системы комплемента.
Первичные ИДС с преимущественным поражением
гуморального адаптивного иммунитета
Болезнь Брутона
Впервые в 1952 году американский педиатр Брутон (Bruton) описал 8-летнего мальчика, страдавшего различными инфекционными заболеваниями, который к 4-летнему возрасту 14 раз болел пневмонией, неоднократно отитами, синуситами, перенес сепсис и менингит. При исследовании у него в сыворотке крови не обнаружили антител. Это было первое описание первичного иммунодефицита в научной медицинской литературе, выделенного как самостоятельная нозологическая форма.
Болезнь Брутона имеет рецессивный тип наследования, сцепленный с Х-хромосомой, болеют ей только мальчики. Заболевание также обозначается как Х-сцепленная агаммаглобулинемия. Встречается с частотой 1/1000000 населения. В Великобритании частота этого иммунодефицита составляет 1/100000.
В основе болезни Брутона лежит дефект фермента цитоплазматической тирозинкиназы, который осуществляет передачу сигнала в ядро В-лимфоцитов для его активации с последующим превращением в плазматическую клетку. Тем самым становится невозможным синтез иммуноглобулинов.
Первые признаки заболевания проявляются начиная с 7-8 месяцев до 2-3 лет. Трансплацентарный перенос иммуноглобулинов обычно обеспечивает больных этой болезнью детей количеством антител, достаточным для того, чтобы избегать инфекций в течение нескольких первых месяцев жизни. В дальнейшем отсутствие синтеза антител приводит, как правило, к возникновению рецидивирующих бактериальных инфекций, преимущественно дыхательных путей и кожи. Обычно возбудителями являются стрептококки, стафилококки и грамотрицательные бактерии. Инфекции имеют тенденцию распространяться, что приводит к септицемии и менингиту. Однако у таких детей сохраняется устойчивость к вирусным инфекциям, так как клеточный иммунитет не нарушен.
При объективном осмотре находят необычно маленькие, гладкие миндалины, лимфатические узлы небольшие, селезенка не увеличена. Обращает особое внимание тот факт, что лимфоузлы, печень и селезенка не реагируют увеличением на инфекционно-воспалительный процесс в организме, что является важным диагностическим признаком.
В слизистой оболочке кишечника полностью исчезают плазматические клетки, хотя в норме они там содержаться в достаточно большом количестве. В связи с этим наблюдаются нарушения всасывания, нередко развиваются хронические энтериты. В кишечнике часто выявляют абсцессы со скоплением распадающихся лейкоцитов в криптах кишки.
При лабораторном обследованиив периферической крови отмечается резкое снижение или отсутствие В-лимфоцитов, низкие уровни или отсутствие всех классов иммуноглобулинов.
Лечение: постоянная заместительная терапия иммуноглобулинами. Введение препаратов иммуноглобулинов производится внутривенно по 200-600 мг/кг в месяц. В случае возникновения инфекционного заболевания дополнительно вводят специфические иммуноглобулины (антистафилококковый, антистрептококковый, противокоревой и т.д.).
Прогноз: при правильно поставленном диагнозе и своевременно начатом лечении относительно благоприятный.
Дата добавления: 2015-01-17; просмотров: 465; Мы поможем в написании вашей работы!; Нарушение авторских прав |