КАТЕГОРИИ:
АстрономияБиологияГеографияДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРиторикаСоциологияСпортСтроительствоТехнологияФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Метод валентных связей
Теория валентных связей (МВС) применительно к комплексным соединениям была разработана Л.Полингом в 1930г. В настоящее время ее используют сравнительно редко, но она прекрасно служила около четверти века химии координационных соединений для объяснения некоторых свойств комплексов (пространственное строение, магнитные свойства). Несмотря на громоздкость количественных расчетов, большие проблемы в интерпретации разнообразных искажений октаэдрических комплексов, отсутствие предсказательной способности даже в случаях высокосимметричного геометрического строения координационных сфер и другие недостатки, МВС остается удобным инструментом, позволяющим наглядно на качественном уровне объяснить факт образования комплексов, дающим возможности оценивать взаимные предпочтения к связыванию, предрасположенность комплексов к гидролизу, поликонденсации, предсказывать состав и некоторые свойства карбонилов и родственных соединений и, конечно, объяснять, а во многих случаях и предсказывать магнитные свойства комплексов.
Основные положения МВС, касающиеся структуры комплексов формулируются следующим образом:
1. Связь между комплексообразователем и лигандами устанавливается по донорно-акцепторному механизму, причем в σ–связи лиганд является донором электронной пары ("кислотой Льюиса"), центральный атом – акцептором ("основанием Льюиса").
2. Мерой прочности связи служит степень перекрывания орбиталей. Для объяснения факта образования прочных связей при вполне конкретном пространственном расположении лигандов вокруг центрального атома, зачастую не совпадающим с пространственным расположением его собственных вакантных АО вводится понятие о гибридизации комплексообразователя, участвующих в σ–связывании. Тип гибридизации определяется числом, природой центрального атома и лигандов. Характер гибридизации определяет геометрическую форму комплекса.
3. Дополнительное упрочение комплекса обусловлено возникновением дополнительного π–связывания. При этом зачастую в качестве донора выступает электроположительный атом комплексообразователя, а акцептором – более электроотрицательный атом, за счет которого координируется лиганд. Такое донорно-акцепторное взаимодействие получило название дативного.
4. Магнитные свойства, проявляемые комплексом, объясняются особенностями заселения электронами орбиталей комплексообразователя. При наличии неспаренных электронов комплекс парамагнитен. Полное отсутствие неспаренных электронов обуславливает диамагнетизм комплексного соединения. Приближенное значение магнитного момента μ (в магнетонах Бора, μВ) можно рассчитать по формуле
 , (4.10)
, (4.10)
где n – число неспаренных электронов.
Прежде чем разобрать несколько примеров применения МВС для анализа строения и свойств ряда комплексов,
 |
полезно вспомнить некоторые сведения об электронном строении, валентных возможностях потенциальных комплексообразователей и лигандов, а также прокомментировать отдельные положения теории Л.Полинга.
Атомы второго периода, выступая в качестве комплексообразователей (Ве, В), а стало быть, устанавливая в заметной степени ковалентные связи с лигандами, ограничены в предельно достижимых КЧ, т.к. на валентном энергетическом уровне имеют только четыре орбиталями (2s– и 2р–). Элементы III-го и больших периодов располагают вакантными nd–орбиталями и за счет них могут проявить повышенные акцепторные свойства (увеличить КЧ до 6 и более, установить дополнительные π–связи с лигандами σ– и π–донорами). Однако, как уже отмечалось ранее (гл. 1.5), энергия nd–орбиталей довольно велика. В то же время их энергетическая выгодность для электронов усиливается при связывании рассматриваемого атома с сильно электроотрицательными элементами (особенно со F–, и лигандами, координирующимися атомами кислорода: О2–, ОН–, ОН2 и т.п.). Впервые предположение о возможности использования в связях внешних d–орбиталей было высказано в 1937г. Хиггинсом, а позднее оно нашло расчетное подтверждение.
Атомы переходных элементов[40] располагают, к тому же еще и (n-1)d–орбиталями, которые гораздо более валентны, чем nd–орбитали, особенно у первых элементов декад, особенно в невысоких положительных степенях окисления. По мере заполнения (n-1)d–орбиталей электронами их акцепторные возможности ослабевают (усиливается вероятность использования в этом качестве nd–орбиталей), зато растут донорные свойства и, соответственно, усиливаются предпочтения к связыванию с лигандами σ–донорами и π–акцепторами.
Чтобы различать два вида комплексов, были введены понятия: внешнеорбитальные и внутриорбитальные (Таубе), спин-свободные и спин-спаренные (Ньюхольм), высокоспиновые и низкоспиновые (Оргел).
Участвующие в ковалентном связывании атомные орбитали должны быть сопоставимы по энергии и соответствовать друг другу по симметрии: располагаться таким образом, чтобы обеспечить перекрывание участками, в которых знаки волновых функций совпадают. Поскольку s–орбиталь среди валентных обычно имеет самую низкую энергию, она практически всегда используется в связывании,[41] но из-за сферической симметрии она не может участвовать в π–перекрывании, а σ–взаимодействие может поддерживать в любом направлении (в том числе, и будучи задействована в процессах гибридизации). Симметрия р–орбиталей позволяет им участвовать как в σ–, так и в π–перекрываниях. В составе центрального атома для поддержания его КЧ (больше единицы: 6, 4, реже другие) р–орбитали предварительно гибридизуются с s– и, при необходимости, с d–орбиталями. Кроме того, симметрия р–орбиталей позволяет им участвовать и в π–связывании (обычно, в составе донорных атомов лигандов). При высоких КЧ (4 и выше) в σ–связывании могут вовлекаться и d–орбитали подходящей симметрии (в квадратах и октаэдрах – расположенные лепестками вдоль прямоугольных осей координат dx2-y2, dz2, а при тетраэдрическом окружении – расположенные по биссектрисам координатных углов dxy, dxz, dyz). По причинам, которые будут пояснены позже, первые две орбитали имеют групповое обозначение dγ (или еg), а три другие – dε (или t2g). Симметрия d–орбиталей позволяет им участвовать и в π–взаимодействии, причем, из-за некоторой направленности в сторону потенциального партнера они могут обеспечить более сильное перекрывание электронных облаков, чем то, что достигается при использовании в π–связях р–орбиталей сопоставимой энергии (близких по размеру).
Таблица 4.11
Форма и относительная прочность гибридных связей (Е*)
| КЧ | Тип гибридизации | Геометрическая форма | Е* |
| – | s | – | 1.000 |
| – | p | – | 1.732 |
| sp | Линия | 1.932 | |
| sp2 | Треугольник | 1.991 | |
| sp3, dε3s | Тетраэдр | 2.000 | |
| dx2-y2sp2 | Квадрат | 2.694 | |
| dz2sp3 | Тригональная бипирамида | – | |
| dx2-y2sp3, dε3dx2-y2s | Квадратная пирамида | – | |
| dε3sp2 | Октаэдр | 2.923 |
Наиболее часто встречающиеся типы гибридизации, соответствующие им (полученные расчетным путем) геометрические формы комплексов, а также относительная прочность σ–связей, образуемых с помощью соответствующих гибридных орбиталей, приведены в таблице 4.11.
Что касается третьего пункта, то поводом для постулирования этого положения стали примеры прочного связывания некоторых 4d– и 5d–элементов с лигандами, донорные свойства которых выражены достаточно слабо. Например, Pt(II), Hg(II), Au(III) лучше связываются с крупными галогенид-ионами, чем с F–; они же образуют достаточно прочные комплексы с :PF3 и ∶P(C6H5)3, но вовсе не связываются с ∶РН3 (напомним, что молекула ∶РН3 очень неохотно связывается с таким активным акцептором электронной пары, как Н+). Эти факты были объяснены Полингом несколькими причинами, одна из которых – растущая кратность связи за счет дополнительного дативного π–взаимодействия комплексообразователей с конфигурациями d8, d10 с d–орбиталями атомов Cl, Br, J, P. В свою очередь d–орбитали фосфора активней вовлекаются в связывание в составе таких лигандов, где их энергия понижена под влиянием собственных внутрилигандных сильно электроотрицательных атомов (F) или группировок (С6Н5).
Существование разнообразных форм дополнительного π–связывания M–L было в дальнейшем подкреплено множеством разнообразных примеров. Важнейшие типы π–взаимодействия в комплексах могут быть систематизированы следующим образом (рис.4.26):
а) πd(M)→p(L): частичный переход электронов с d–орбитали металла на вакантные р–орбитали лиганда;
б) πd(M)→d(L): частичный переход электронов с d–орбитали металла на вакантные d–орбитали лиганда;
в) πp(M)←p(L): частичный переход электронов с р–орбитали лиганда на вакантные р–орбитали металла;
г) πd(M)←p(L): частичный переход электронов с р–орбитали лиганда на вакантные d–орбитали металла.

| М: d–элементы, замыкающие вставные декады и находящиеся в низких степенях окисления, р–элементы больших периодов; L: CO, CN–, NO2–, …(a); L: Cl–, Br–, J–, S2–, PR3…(б) |
| М: Ве, В (в); d–элементы, возглавляющие вставные декады и находящиеся в высших степенях окисления и р–элементы III-го и последующих периодов в высших положительных степенях окисления (г); L: О2–, ОН–, ОН2, F–, Cl–… |

| Рис.4.26 – Способы образования π–связей M–L. (Орбитали изображены в состоянии до связывания, затемненными показаны орбитали донора). |
Теперь можно закрепить применение разобранных положений теории Полинга на конкретных примерах, при их анализе рассмотрим и магнитные свойства комплексов. Вначале обсудим состав, структуру и некоторые свойства комплексных соединений d–металлов.
Для первых d–элементов характерны высшие положительные степени окисления. Это формально означает, что в качестве комплексообразователя выступает полностью ионизированный атом, имеющий много пустых орбиталей и, соответственно, он должен предпочтительно связываться с лигандами σ– и π–донорами. В частности, для самыми стабильными комплексами Ti4+являются фторидный (в меньшей степени – другие галогенидные) и кислородсодержащие. Если не принимать во внимание полимерные соединения, то это анионный комплекс [TiF6]2– и катионный [Ti(ОН)2(ОН2)4]2+ (аквокомплекс "[Ti(ОН2)6]4+ " очень сильно гидролизуется под сильным поляризующим воздействием центрального атома; в степени окисления +III аквокомплекс гидролизуется в гораздо меньшей степени: [Ti(ОН2)6]3+). Электронная конфигурация Ti4+: [Ar]3d04s04p0, в σ–связывании с лигандами участвуют d2sр3-гибридные орбитали, пустые dε могут быть задействованы в дополнительном многоцентровом πd(M)←p(L)–связывании:

Катионные комплексы Ti4+и Ti3+также являются внутриорбитальными, имеют октаэдрическую симметрию, но в отличие от [Ti(ОН2)6]3+(и [TiF6]2–) дигидроксо-диаквотитан (IV) имеет искаженную структуру: связи с гидродроксо-группами короче, чем с молекулами воды (КЧ = 2+4). Это можно объяснить неравноценным π–связыванием (более сильными π–донорными свойствами ионов ОН–). В то же время [Ti(ОН2)6]3+является парамагнитной частицей, тогда, как [TiF6]2–и [Ti(ОН)2диамагнитны, что согласуется с электронными конфигурациями центрального атома (d1и d0 у Ti3+ и Ti4+, соответственно). Однако теория Полинга не могла объяснить "бросающегося в глаза" различия в окрашенности: комплексы Ti4+бесцветны, а все комплексы Ti3+имеют красно-фиолетовую окраску.
В ряду d–элементов с ростом заряда ядра увеличиваются энергии ионизации атомов, в особенности полная потеря валентных электронов и достижение высшей положительной степени окисления. В частности, атом хрома наиболее стабильные и разнообразные комплексы образует в степени окисления +III. Электронная конфигурация Cr3+– d3, что позволяет, опираясь на закономерности электронного строения атомов и идеи МВС, сделать некоторые прогнозы/выводы:
· даже, если все электроны будут распределены в соответствии с правилом Хунда, на 3d–подуровне Cr3+остаются две вакантные dγ–орбитали, что сохраняет возможность для образования внутриорбитальных октаэдрических комплексов (вполне достижимо КЧ=6);
(Cr3+образует только октаэдрические комплексы)
· т.к. dε–орбитали заселены электронами комплексообразователя, то πd(M)←p(L)–связывание затруднено и образование комплексов с лигандами σ– и π–донорами должно быть более вероятным, чем для Ti4+;
(в водных растворах при высокой концентрации соответствующих лигандов могут быть получены цианидный и даже смешанные аквохлоридные комплексы: [Cr(CN)6]3–, [Cr(ОН2)5Cl]2+и [Cr(ОН2)4Cl2]+, однако данные частицы (особенно последние) легко вступают в реакции замещения на F–или (менее охотно) на кислородсодержащие лиганды:
[CrF6]3–, [Cr(ОН2)6]3+, [Cr(ОН)6]3–)
· все комплексы Cr3+должны быть парамагнитными, т.к. комплексообразователь располагает тремя электронами;
(магнитные моменты всех комплексов Cr3+соответствуют наличию
трех неспаренных электронов).
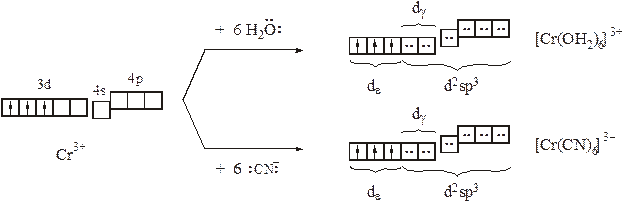
Отметим, что из-за частичной заселенности dε–орбиталей, Cr3+ не может проявить ни π–акцепторных свойств (в составе [Cr(ОН2)6]3+), ни π–донорных (в составе [Cr(CN)6]3–). Любопытно, что цианидные комплексы (карбонилы и другие комплексы с лигандами активными π–акцепторами) нередко проявляют высокое сродство к электрону, что позволяет в составе таких соединений стабилизировать у d-элементов аномально низкие (порой даже отрицательные) степени окисления. В частности К3[Cr(CN)6] по реакции с атомарным водородом (цинк в солянокислой среде) удается восстановить до К6[Cr(CN)6]. Причем в составе нового комплекса атом хрома принимает три дополнительных электрона на свои орбитали и, приобретая нулевую степень окисления, должен был бы, тем самым, воспроизвести электронную конфигурацию нейтрального атома [Ar]3d54s14p0с шестью неспаренными электронами. Однако комплекс К6[Cr(CN)6] диамагнитен. Подобные факты дали основания предположить, что в комплексах с активными π–акцепторами меняется электронное строение комплексообразователя: на d-подуровне в первую очередь заселяются dε–орбитали (поначалу в соответствие с правилом Хунда, а при конфигурациях d4, d5и d6 – попарно). Это позволяет, во-первых, сохранять (n-1)dγ–орбитали вакантными и использовать их для внутриорбитальной гибридизации и σ–связывания, а во-вторых, попарно заполненные dε–орбитали могут быть задействованы для дополнительного πd(M)→p(L)–взаимодействия, что приводит к увеличению кратности связи комплексообразователь–лиганд. Принимая во внимание эти рассуждения, образование комплекса [Cr(CN)6]6–с точки зрения МВС может быть схематично показано следующим образом:

Особенность цианид-ионов в качестве лигандов подтверждает и сравнение комплексов хрома (II): при одинаковом электронном строении центрального атома (d4) магнитные моменты [Cr(CN)6]4–, с одной стороны, и [Cr(ОН2)6]2+, [CrF6]4–, [Cr(NCS)6]4–,…, с другой, отличаются:
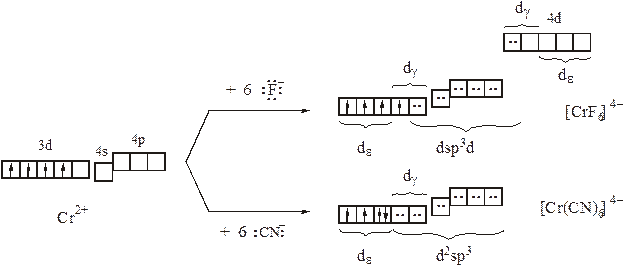
В то же время МВС оказывается бессилен перед объяснением различий в оптических свойствах (окрашенности) и деталей пространственного строения: в отличие от цианидного комплекса все прочие, несмотря на однородный лигандный состав и равноценность участвующих в σ–связывании гибридизованных орбиталей центрального атома, характеризуются слабым тетрагональным искажением октаэдрической координации (КЧ = 4+2).
При дальнейшем повышении заряда ядра и одновременном увеличении числа электронов на валентных орбиталях наблюдается:
ü растущая стабилизация низких степеней окисления d–элементов;
ü усиление π–донорных свойств атомов (ионов) d–металлов. Соответственно постепенно ослабевает взаимодействие с лигандами σ– и π–донорами, растет предпочтение к связыванию с лигандами π–акцепторами, как следствие – комплексы становятся более разнообразными;
ü постепенный переход к внешнеорбитальным комплексам.
Рассмотрим некоторые комплексы Ni (II), Cu (II) и Cu (I).
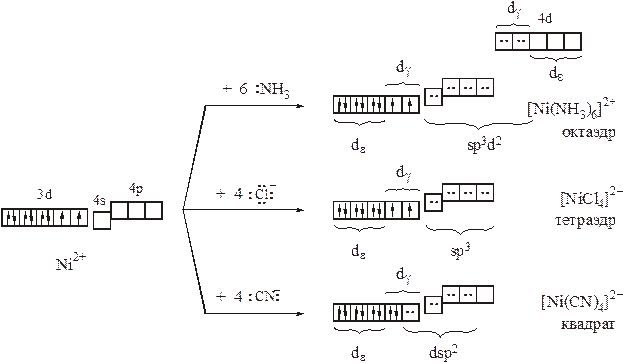
Комплексы Cu (II) весьма разнообразны по лигандному составу: перечень только монодентантных лигандов, при связывании с которыми могут быть получены островные комплексы, включает в себя Н2О, OH–, Г–, NH3, SCN–, S2O32–, NO2–и т.д. Весьма разнообразна их окраска: голубые, желто-зеленые, сине-фиолетовые,… . В то же время магнитные свойства комплексов одинаковы, а их структуры сходны или родственны:
– при электронной конфигурации центрального атома d9во всех комплексах иона Cu2+ обнаруживается один неспаренный электрон;
– в большинстве комплексов реализуется тетрагонально искаженная октаэдрическая координация (КЧ = 4+2); порой оба или один из слабо связанных лигандов полностью покидают координационную сферу (при этом получаются или квадратные – КЧ=4 (нет тетраэдров!), или квадратно-пирамидальные комплексы – КЧ=4+1):
| КЧ = 4+2 (вытянутый октаэдр) | КЧ = 4+1 (квадратная пирамида) | КЧ = 4 (квадрат) |
| [Cu(ОН2)6]2+, [Cu(OH)6]4–, [Cu(NН3)6]2+, [Cu(NН3)4(ОН2)2]2+, [Cu(SCN)6]4– | [CuCl5]3–, [Cu(NН3)4(ОН2)]2+ | [CuCl4]2–, [Cu(NН3)4]2+, [Cu(NО2)4]2–, [Cu(OH)4]2– |
С точки зрения МВС все комплексы Cu (II) являются внешнеорбитальными:
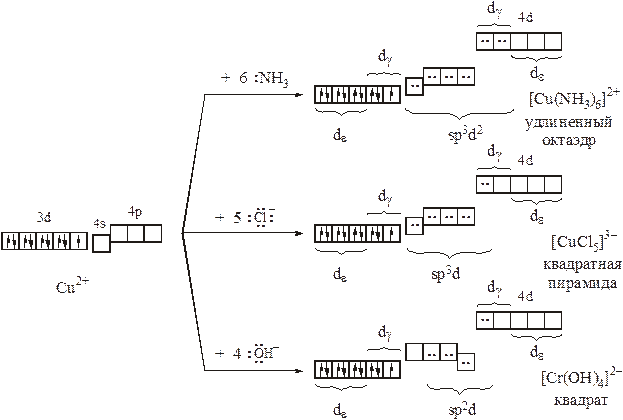
Напомним, что для формирования электронных облаков, ориентированных к вершинам квадратной пирамиды, в гибридизацию должна вовлекаться орбиталь dx2-y2. Она же необходима для образования плоско-квадратных комплексов, в то время как рz–орбиталь из гибридизации извлекается. Кроме того, следует отметить, что, в соответствие с МВС, в хлоридном и гидроксокомплексе возможно слабое дополнительное π–связывание (ионы Cl–являются слабыми π–донорами и π–акцепторами; ионы ОН–обладают гораздо более выраженными π–донорными свойствами, но центральный атом π–акцепторные свойства может реализовать только за счет высоколежащих 4d–орбиталей). Несмотря на объяснения способов ковалентного взаимодействия центрального атома и лигандов, МВС, по-прежнему, бессилен предложить причины, как спектральной активности, так и структурных особенностей комплексов. Любопытно, что комплексы Cu (I) наоборот, в подавляющем большинстве бесцветны, но гораздо более разнообразны в структурном плане, несмотря на более низкие координационные числа (КЧ: 2, 3, 4; координационные формы: линия, треугольник, тетраэдр – нет квадратов!):
| КЧ = 2 (линия) sp-гибридизация | КЧ = 3 (треугольник) sp2-гибридизация | КЧ = 4 (тетраэдр) sp3-гибридизация |
| [Cu(NН3)2]+, [CuГ2]–, [Cu(CN)2]– (островная форма) | [CuГ3]2–, [Cu(CN)2]– (цепочечный полимер – [Cu(CN)2/2(CN)]–) | [Cu(CN)4]3–, [Cu(SCN)4]3–, [CuCl4]3– (искаж. тетр.) |
Что касается комплексов s– и р–элементов, то кратко отметим лишь некоторые важные закономерности:
· В качестве комплексообразователей выступают ионы элементов (см. табл.4.7) с промежуточным поляризующим действием (электро-отрицательностью), однако важно понимать, что у большинства рассматриваемых элементов эти характеристики заметно выше, чем у d–металлов;
· Практически все потенциальные комплексообразователи образуют только октаэдрические комплексы (у Ве2+, В3+известны только тетраэдры; Al3+ и Ga3+наряду с октаэдрами тоже порой образуют тетраэдрические комплексы; Sn2+, Pb2+имеют только тетраэдрические и тригонально-пирамидальные комплексы), что требует вовлечение в гибридизацию и σ–взаимодействие ndγ-орбиталей (за счет s– и р–орбиталей может быть реализовано только КЧ=4). Это предполагает связывание с сильно электроотрицательными атомами, а также то, что за счет вакантных ndε-орбиталей потенциальные комплексообразователи являются достаточно активными π–акцепторами.
· В качестве лигандов в подавляющем большинстве случаев выступают активные σ– и π–доноры: ОН2 (только при связывании с ионами, не вызывающими сильный гидролиз, т.е. п/д, которых минимально в данном ряду элементов), ОН–(при связывании с ионами, характеризующимися промежуточным уровнем п/д в ряду данных элементов), одноатомные лиганды: О2–, F–. Р–элементы VI–го, V–го и, в меньшей степени, IV–го периодов имеют заполненные (n-1)d10–подуровни и, поэтому могут участвовать в πd(M)→d(L)–взаимодействии. Соответственно, для таких элементов даже в водной среде могут оказаться вполне конкурентными, выгодными связи М–Cl и Cl– в качестве потенциального лиганда. В ряде случаев стабилизируются комплексы и с более крупными галогенами. Те же элементы, но гораздо реже могут образовать островные воднорастворимые комплексы с лигандами S2–и SH–.
· Все комплексы р–элементов диамагнитны и в подавляющем большинстве своем – бесцветны. Чрезвычайно редкие исключения возможны в случае комплексов с лигандами π–акцепторами.
Таблица 4.12
Составы важнейших островных
воднорастворимых комплексов р–элементов
| IIа | IIIа | IVа | Vа | VIа |
| [Be(OH2)4]2+[Be(OH)4]2–[BeF4]2– | [B(OH)4]–[B4O5(OH)4]2– [BF4]– | –– | –– | –– |
| [Mg(OH2)6]2+ | [Al(OH2)6]3+ [Al(OH)4]– [Al(OH)4(OH2)2]–[Al(OH)6]3– [AlF6]3– | [SiO2(OH2)2]2– [SiF6]2– | [PF6]– | –– |
| То же, что у Al3+ | [Ge(OH)4]2–[GеO2(OH)2]2–[GeF6]2– | [AsF6]– AsO43–; [As(OH)4]– [AsF4]– [AsCl4]– | –– | |
| То же, что у Al3+, кроме гидроксокомплекса, дополнительно [InCl4]– [InCl6]3– | [Sn(OH)2(OH2)4]2+ [Sn(OH)6]2– [SnF6]2– [SnCl6]2–; [Sn(OH2)6]2+ [SnCl3]– [Sn(OH)3]– | [Sb(OH)6]– [SbF6]– [SbCl6]–; [Sb(OH)2(OH2)2]+ [Sb(OH)4]– [Sb(OH)6]3–[SbCl4]– | [Te(OH)6]; [Te(OH)6]2– [TeCl6]2– | |
| [TlCl6]3–; [TlCl3]2– | [Pb(OH)6]2– [SnF6]2–; [Pb(OH2)6]2+ [PbCl3]– [Pb(OH)3]– | [Bi(OH2)6]3+ [BiCl4]– | [Po(OH)2(OH2)4]2+ [Ро(OH)6]2– [РоCl6]2–; [Ро(OH2)6]2+ |
В заключение кратко обсудим применение идей МВС для объяснения состава, структуры и некоторых свойств достаточно своеобразных соединений: карбонилов и карбонильных комплексов d–элементов (известны также и полилигандные карбонилы: карбонилнитрозилы (M(CO)x(NO)y), карбонилгалогениды (M(CO)xГy), карбонилгидриды (M(CO)xHy), карбонил-металлоцены (M(CO)x(C5H5)y) и т.п., в том числе, полиядерные, содержащие несколько атомов d–металла). Состав большинства из них подчиняется правилам, сформулированным в 20-е годы ХХв. на рубеже становления квантово-механической модели строения атома: первое и модифицированное правило Сиджвика (правило 18 электронов): наиболее стабильными являются комплексы, в составе которых центральный атом имеет полностью завершенную (n-1)d10ns2np6-конфигурацию. В расчет принимаются валентные электроны d–элемента и электроны лигандов, задействованные в связях M–L. Правило основано на предположении попарного заселения валентных орбиталей электронами центрального атома и донорно-акцепторном взаимодействии комплексообразователь–лиганд (лиганды-радикалы, типа NO, рассматриваются как доноры трех электронов; лиганды с протяженными π–системами являются донорами всех своих π–электронов).
Таблица 4.13
Состав известных карбонилов 3d-элементов
| Vв | VIв | VIIв | VIIIв | ||
| V(CO)6 "V2(CO)12" очень нестабилен | Cr(CO)6 | Mn2(CO)10 | Fe(CO)5 Fe2(CO)9 Fe3(CO)9 | Co2(CO)8 Co4(CO)12 | Ni(CO)4 |
Объем и тематика данного учебника не позволяют выполнить анализ возможных причин, ограничивающих круг элементов, склонных к образованию карбонилов (табл.4.14). Отметим только, что с учетом родственных соединений они получены для всех d-металлов за исключением
Таблица 4.14
Круг d-элементов, входящих в состав карбонилов
| Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn |
| Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd |
| La | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg |
Nb, Ta, а также элементов подгрупп скандия и цинка. В то же время состав и структуры простейших карбонилов идеально согласуются с правилом Сиджвика и теорией Полинга. В частности, чередование мономерных (у Cr, Fe и Ni) и димерных молекул (у V, Mn и Со) есть результат того, что элементы нечетных групп имеют нечетное число валентных электронов, поэтому мономерные молекулы являются радикалами и способны объединяться за счет связи М–М (такие соединения принято называть кластерами):
ü примеры боснование состава на основе модифицированного правила Сиджвика:
| Электронная конфигурация изолированного атома Mn: [Ar]3d54s24p0 Электронная конфигурация комплексообразователя: [Ar]3d74s04p0 Число электронов на орбиталях центрального атома в составе комплекса: 7 + 5·2(СО) + 1(Mn) = 18 | Электронная конфигурация изолированного атома Fe: [Ar]3d64s24p0 Электронная конфигурация комплексообразователя: [Ar]3d84s04p0 Число электронов на орбиталях центрального атома в составе комплекса: 8 + 5·2(СО) = 18 |
ü структуры на основе теории Полинга

КЧCr = 6 КЧFe = 5 КЧNi = 4
октаэдр тригональная тетраэдр
бипирамида
d2sр3 dsр3 sр3

КЧMn = 6 КЧСо = 4+1
октаэдр тригональная
бипирамида
d2sр3 dsр3
У Fe, Co и некоторых тяжелых d–металлов известны "сложные карбонилы". Убедительных объяснений их состава и избирательного существования, пока не выработано. В то же время особенности их структуры (наличие связей М–М, число мостиковых или концевых молекул СО, пространственное окружение) можно предвидеть, применяя теорию Сиджвика/Полинга (см., например, учебник Дж.Хьюи "Неорганическая химия. Строение вещества и реакционная способность").
Дата добавления: 2014-11-13; просмотров: 1720; Мы поможем в написании вашей работы!; Нарушение авторских прав |